Contribution of the Oligomeric State to the Thermostability of Isoenzyme 3 from Candida rugosa


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Lipases Purification
2.3. Size Exclusion Chromatography (SEC)
2.4. Enzyme Kinetic Assays
2.5. Other Methods
2.6. Effect of Temperature and pH on the Stability of C. rugosa Isoenzyme 3
2.7. Statistical Analysis
3. Results
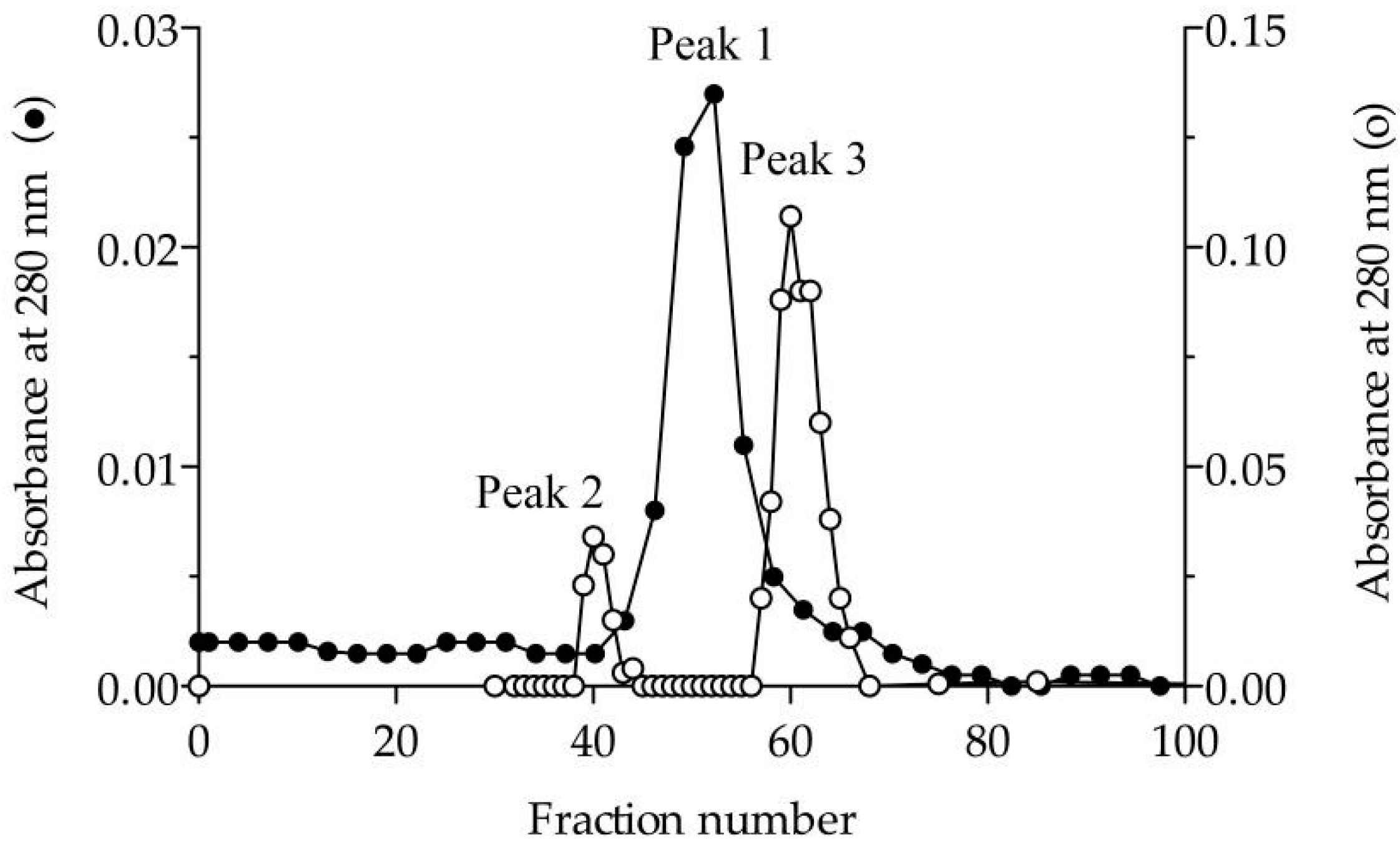
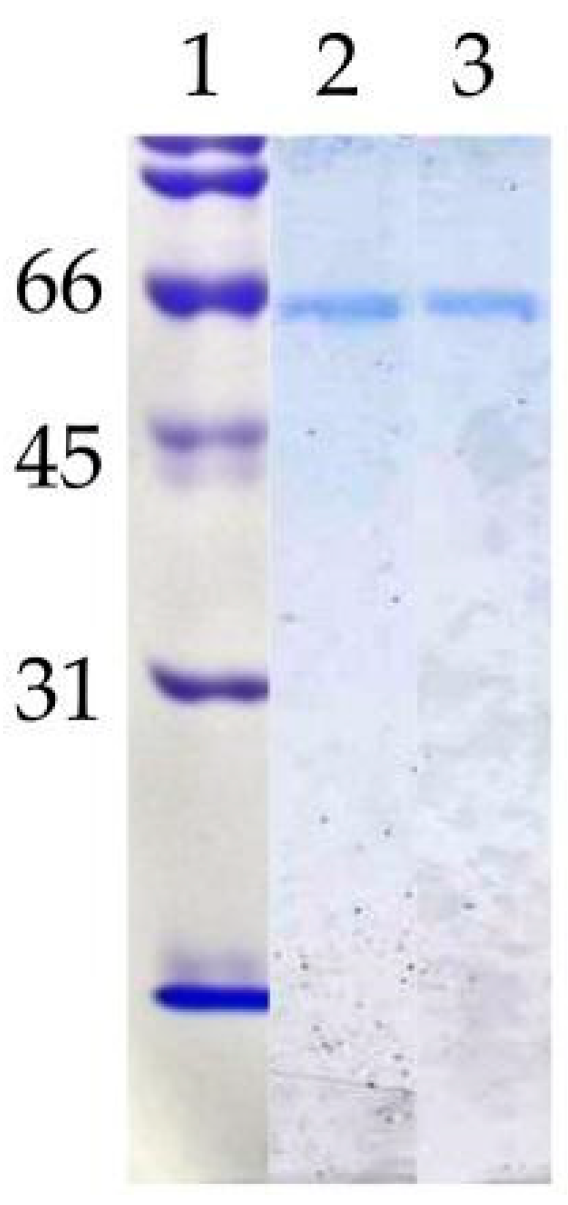
3.1. Purification and Molecular Characterisation
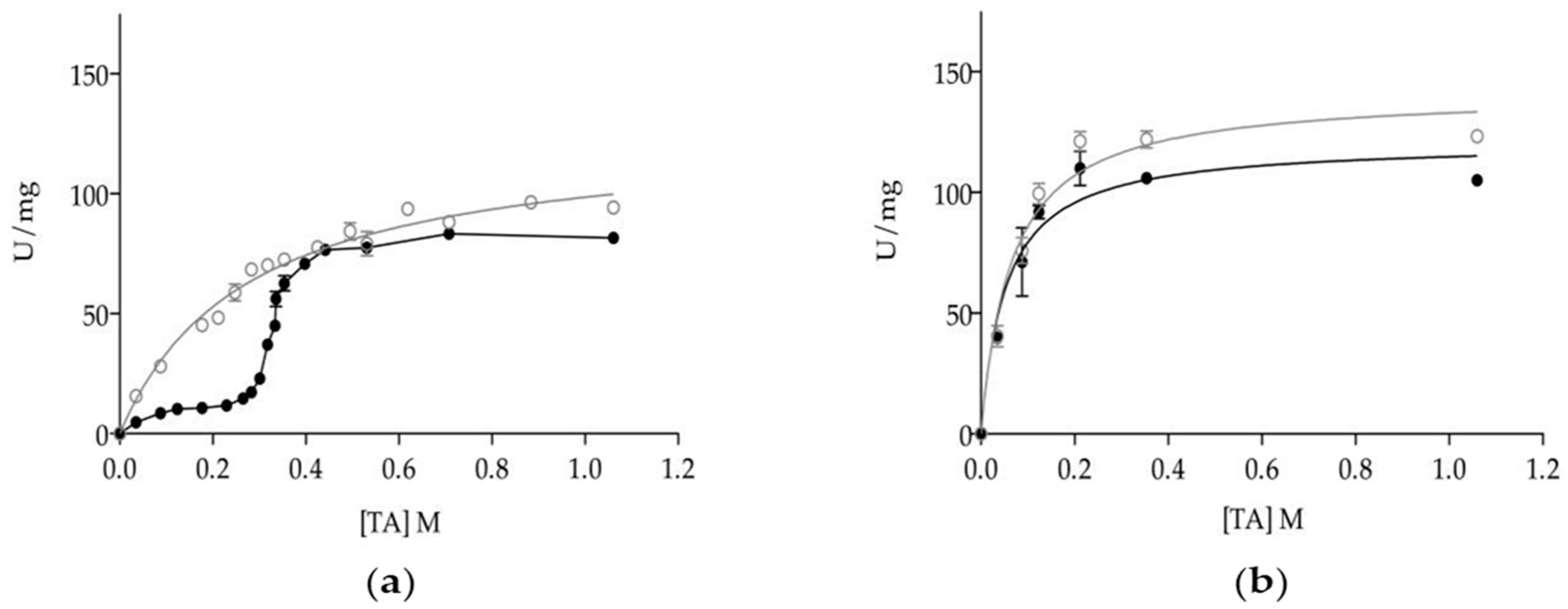
3.2. Effect of Oligomerization on Enzyme Kinetics
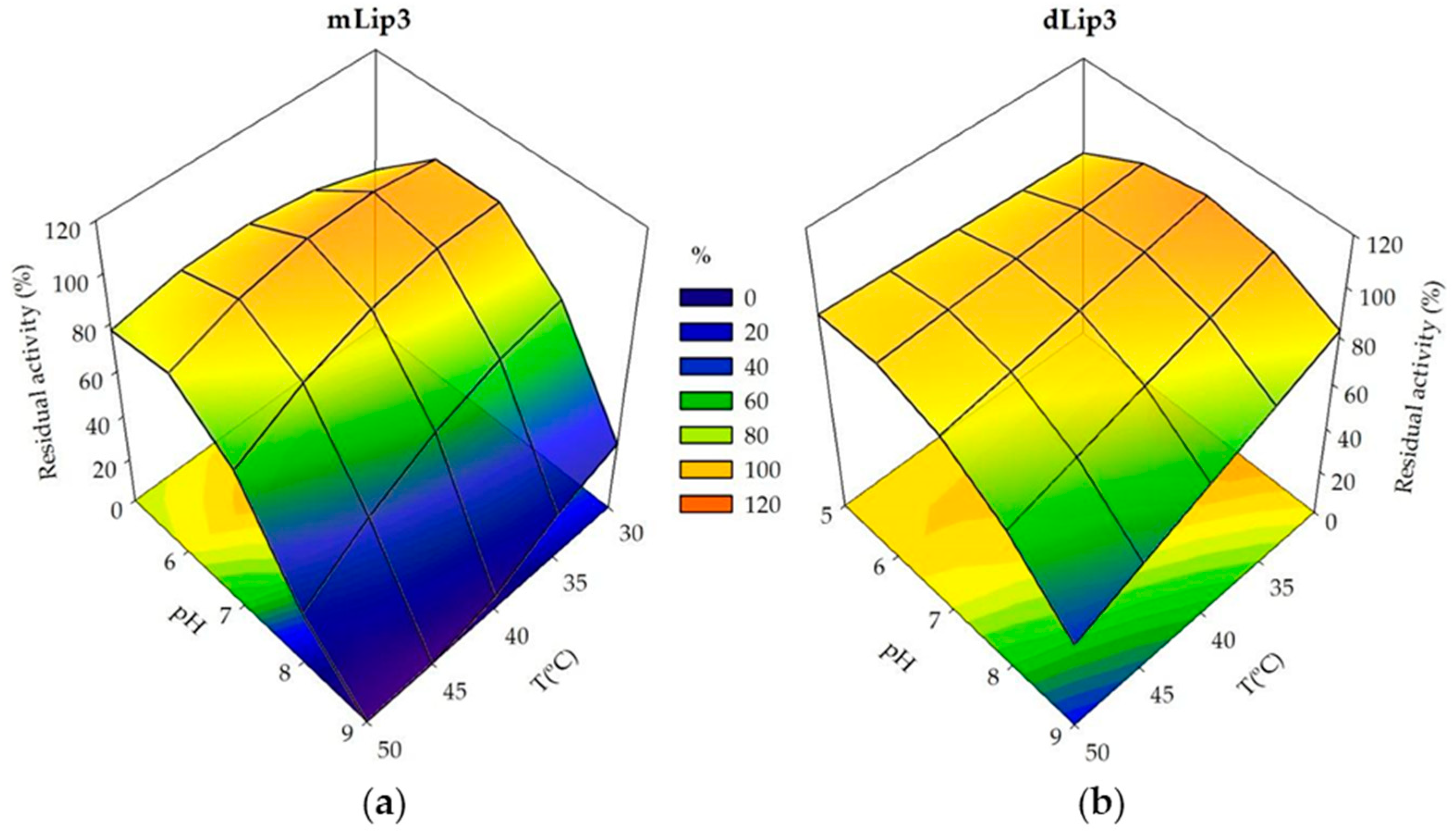
3.3. Effect of Dimerization on Enzyme Thermostability
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Reed, C.J.; Lewis, H.; Trejo, E.; Winston, V.; Evilia, C. Protein adaptations in archaeal extremophiles. Archaea 2013, 2013, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, M.C.; Tron, P.; Leroy, G.; Aubert, C.; Tauc, P.; Giudici-Orticoni, M.T. A new sulfurtransferase from the hyperthermophilic bacterium Aquifex aeolicus: Being single is not so simple when temperature gets high. FEBS J. 2007, 274, 4572–4587. [Google Scholar] [CrossRef] [PubMed]
- Devenish, S.R.A.; Gerrard, J.A. The role of quaternary structure in (β/α)8-barrel proteins: Evolutionary happenstance or a higher level of structure-function relationships? Org. Biomol. Chem. 2009, 7, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Linde, M.; Heyn, K.; Merkl, R.; Sterner, R.; Babinger, P. Hexamerization of geranylgeranylglyceryl phosphate synthase ensures structural integrity and catalytic activity at high temperatures. Biochemistry 2018, 57, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Loveridge, E.J.; Rodriguez, R.J.; Swanwick, R.S.; Allemann, R.K. Effect of dimerization on the stability and catalytic activity of dihydrofolate reductase from the hyperthermophile Thermotoga maritima. Biochemistry 2009, 48, 5922–5933. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.-S.; Rhee, J.-K.; Kim, N.D.; Yoon, J.; Kim, U.; Koh, E.; Oh, J.-W.; Cho, H.-S. Crystal structure of hyperthermophilic esterase EstE1 and the relationship between its dimerization and thermostability properties. BMC Struct. Biol. 2007, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, P.-Y.; Chen, X.-L.; Ji, P.; Li, C.-Y.; Wang, P.; Zhang, Y.; Xie, B.-B.; Qin, Q.-L.; Su, H.-N.; Zhou, B.-C.; et al. Interdomain hydrophobic interactions modulate the thermostability of microbial esterases from the Hormone-ensitive Lipase family. J. Biol. Chem. 2015, 290, 11188–11198. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Shivakumaraswamy, S.; Gummadi, S.N.; Manoj, N. Role of an N-terminal extension in stability and catalytic activity of a hyperthermostable α/β hydrolase fold esterase. Protein Eng. Des. Sel. 2017, 30, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Perica, T.; Kondo, Y.; Tiwari, S.P.; McLaughlin, S.H.; Kemplen, K.R.; Zhang, X.; Steward, A.; Reuter, N.; Clarke, J.; Teichmann, S.A. Evolution of oligomeric state through allosteric pathways that mimic ligand binding. Science 2014, 346, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fraser, N.J.; Liu, J.; Mabbitt, P.D.; Correy, G.J.; Coppin, C.W.; Lethier, M.; Perugini, M.A.; Murphy, J.M.; Oakeshott, J.G.; Weik, M.; et al. Evolution of protein quaternary structure in response to selective pressure for increased thermostability. J. Mol. Biol. 2016, 428, 2359–2371. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Hashimoto, K.; Madej, T.; Panchenko, A.R. Evolutionary, physicochemical, and functional mechanisms of protein homooligomerization. Prog. Mol. Biol. Transl. Sci. 2013, 117, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Elgharbi, F.; Ben Hlima, H.; Ameri, R.; Bejar, S.; Hmida-sayari, A. A trimeric and thermostable lichenase from B. pumilus US570 strain: Biochemical and molecular characterization. Int. J. Biol. Macromol. 2017, 95, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Teichmann, S.A. Protein flexibility facilitates quaternary structure assembly and evolution. PLoS Biol. 2014, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Sharma, C. Glycine-rich loop encompassing active site at interface of hexameric M. tuberculosis Eis protein contributes to its structural stability and activity. Int. J. Biol. Macromol. 2018, 109, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Aschauer, P.; Rengachari, S.; Lichtenegger, J.; Schittmayer, M.; Padmanabha Das, K.M.; Mayer, N.; Breinbauer, R.; Birner-Gruenberger, R.; Gruber, C.C.; Zimmermann, R.; et al. Crystal structure of the Saccharomyces cerevisiae monoglyceride lipase Yju3p. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2016, 1861, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Veeraragavan, K.; Gibbs, B.F. Detection and partial purification of two lipases from Candida rugosa. Biotechnol. Lett. 1989, 11, 345–348. [Google Scholar] [CrossRef]
- Tomizuka, N.; Ota, Y.; Yamada, K. Studies on lipase from Candida cylindracea. Agric. Biol. Chem. 1966, 30, 576–584. [Google Scholar] [CrossRef]
- Rúa, M.L.; Díaz-Mauriño, T.; Fernández, V.M.; Otero, C.; Ballesteros, A. Purification and characterization of two distinct lipases from Candida cylindracea. BBA-Gen. Subj. 1993, 1156, 181–189. [Google Scholar] [CrossRef]
- Rúa, M.L.; Ballesteros, A. Rapid purification of two lipase isoenzymes from Candida rugosa. Biotechnol. Tech. 1994, 8, 21–26. [Google Scholar] [CrossRef]
- Pernas, M.A.; Pastrana, L.; Fuciños, P.; Rúa, M.L. Regulation of the interfacial activation within the Candida rugosa lipase family. J. Phys. Org. Chem. 2009, 22, 508–514. [Google Scholar] [CrossRef]
- Pernas, M.A.; López, C.; Rúa, M.L.; Hermoso, J. Influence of the conformational flexibility on the kinetics and dimerisation process of two Candida rugosa lipase isoenzymes. FEBS Lett. 2001, 501, 87–91. [Google Scholar] [CrossRef]
- López, N.; Pernas, M.A.; Pas trana, L.M.; Sánchez, A.; Valero, F.; Rúa, M.L. Reactivity of pure Candida rugosa lipase isoenzymes (Lip1, Lip2, and Lip3) in aqueous and organic media. Influence of the isoenzymatic profile on the lipase performance in organic media. Biotechnol. Prog. 2004, 20, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.W.; Shieh, C.J.; Lee, G.C.; Shaw, J.F. Multiple mutagenesis of the Candida rugosa LIP1 gene and optimum production of recombinant LIP1 expressed in Pichia pastoris. Appl. Microbiol. Biotechnol. 2005, 67, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-W.; Lee, G.-C.; Shaw, J.-F. Codon optimization of Candida rugosa LIP1 gene for improving expression in Pichia pastoris and biochemical characterization of the purified recombinant Lip1 lipase. J. Agric. Food Chem. 2006, 54, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, J.; Deng, R.; Wang, X. Scale-up fermentation of recombinant Candida rugosa lipase expressed in Pichia pastoris using the GAP promoter. J. Ind. Microbiol. Biotechnol. 2008, 35, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-W.; Lee, G.-C.; Shaw, J.-F. Efficient production of active recombinant Candida rugosa Lip3 lipase in Pichia pastoris and biochemical characterization of the purified enzyme. J. Agric. Food Chem. 2006, 54, 5831–5838. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, P.; Alarcón, M.; Ramón, R.; Dolors Benaiges, M.; Valero, F. Recombinant Candida rugosa LIP2 expression in Pichia pastoris under the control of the AOX1 promoter. Biochem. Eng. J. 2009, 46, 271–277. [Google Scholar] [CrossRef]
- Lee, G.-C.; Lee, L.-C.; Sava, V.; Shaw, J.-F. Multiple mutagenesis of non-universal serine codons of the Candida rugosa LIP2 gene and biochemical characterization of purified recombinant Lip2 lipase overexpressed in Pichia pastoris. Biochem. J. 2002, 366, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.C.; Chen, Y.T.; Yen, C.C.; Chiang, T.C.-Y.; Tang, S.-J.; Lee, G.-C.; Shaw, J.-F. Altering the substrate specificity of Candida rugosa Lip4 by engineering the substrate-binding sites. J. Agric. Food Chem. 2007, 55, 5103–5108. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.-C.; Yen, C.-C.; Malmis, C.C.; Chen, L.-F.; Chen, J.-C.; Lee, G.-C.; Shaw, J.-F. Characterization of codon-optimized recombinant Candida rugosa Lipase 5 (Lip5). J. Agric. Food Chem. 2011, 59, 10693–10698. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-J.; Sun, K.-H.; Sun, G.-H.; Chang, T.-Y.; Lee, G.-C. Recombinant expression of the Candida rugosa Lip4 lipase in Escherichia coli. Protein Expr. Purif. 2000, 20, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-J.; Shaw, J.-F.; Sun, K.-H.; Sun, G.-H.; Chang, T.-Y.; Lin, C.-K.; Lo, Y.-C.; Lee, G.-C. Recombinant expression and characterization of the Candida rugosa Lip4 lipase in Pichia pastoris: Comparison of glycosylation, activity, and stability. Arch. Biochem. Biophys. 2001, 387, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.-C.; Malmis, C.C.; Lee, G.-C.; Lee, L.-C.; Shaw, J.-F. Site-specific saturation mutagenesis on residues 132 and 450 of Candida rugosa Lip2 enhances catalytic efficiency and alters substrate specificity in various chain lengths of triglycerides and esters. J. Agric. Food Chem. 2010, 58, 10899–10905. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Honda, H.; Taniguchi-Morimura, J.; Iwasaki, S. The codon CUG is read as serine in an asporogenic yeast Candida cylindracea. Nature 1989, 341, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Barriuso, J.; Vaquero, M.E.; Prieto, A.; Martínez, M.J. Structural traits and catalytic versatility of the lipases from the Candida rugosa-like family: A review. Biotechnol. Adv. 2016, 34, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Verger, R. Enzyme kinetics of lipolysis. In Methods in Enzymology; Academic Press, Inc.: Salt Lake City, UT, USA, 1980; Volume 64, pp. 340–392. ISBN 9780121819644. [Google Scholar]
- Schrag, J.D.; Cygler, M. Lipases and alpha/beta hydrolase fold. Methods Enzymol. 1997, 284, 85–107. [Google Scholar] [CrossRef] [PubMed]
- Mancheño, J.M.; Pernas, M.A.; Martínez, M.J.; Rúa, M.L.; Hermoso, J.A. Structural insights into the lipase/esterase behavior in the Candida rugosa lipases family: Crystal structure of the Lipase 2 isoenzyme at 1.97 Å resolution. J. Mol. Biol. 2003, 332, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Bouthillier, F.; Smith, P.; Harrison, D.; Rubin, B.; Cygler, M. Insights into interfacial activation from an open structure of Candida rugosa lipase. J. Biol. Chem. 1993, 268, 12843–12847. [Google Scholar] [PubMed]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Cygler, M. Two conformational states of Candida rugosa lipase. Protein Sci. 1994, 3, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Mancheño, J.M.; Pernas, M.A.; Rúa, M.L.; Hermoso, J.A. Crystallization and preliminary X-ray diffraction studies of two different crystal forms of the Lipase 2 isoform from the yeast Candida rugosa. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, D59, 499–501. [Google Scholar] [CrossRef]
- Pletnev, V.; Addlagatta, A.; Wawrzak, Z.; Duax, W. Three-dimensional structure of homodimeric cholesterol esterase-ligand complex at 1.4 Å resolution. Acta Crystallogr.-Sect. D Biol. Crystallogr. 2003, 59, 50–56. [Google Scholar] [CrossRef]
- Ghosh, D.; Wawrzak, Z.; Pletnev, V.Z.; Li, N.; Kaiser, R.; Pangborn, W.; Jornvall, H.; Erman, M.; Duax, W.L. Structure of uncomplexed and linoleate-bound Candida cylindracea cholesterol esterase. Structure 1995, 3, 279–288. [Google Scholar] [CrossRef]
- Kaiser, R.; Erman, M.; Duax, W.L.; Ghosh, D.; Jörnvall, H. Monomeric and dimeric forms of cholesterol esterase from Candida cylindracea. Primary structure, identity in peptide patterns, and additional microheterogeneity. FEBS Lett. 1994, 337, 123–127. [Google Scholar] [CrossRef]
- Pernas, M.; López, C.; Prada, A.; Hermoso, J.; Rúa, M.L. Structural basis for the kinetics of Candida rugosa Lip1 and Lip3 isoenzymes. Colloid Surf. B Biointerfaces 2002, 26, 67–74. [Google Scholar] [CrossRef]
- Pernas, M.A.; López, C.; Pastrana, L.; Rúa, M.L. Purification and characterization of Lip2 and Lip3 isoenzymes from a Candida rugosa pilot-plant scale fed-batch fermentation. J. Biotechnol. 2000, 84, 163–174. [Google Scholar] [CrossRef]
- Ferrato, F.; Carriere, F.; Sarda, L.; Verger, R. A critical reevaluation of the phenomenon of interfacial activation. Methods Enzymol. 1997, 286, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.J.; Lewis, A. Protein measurement with the folin phenol reagent. Readings 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Varejão, N.; De-Andrade, R.A.; Almeida, R.V.; Anobom, C.D.; Foguel, D.; Reverter, D. Structural mechanism for the temperature-dependent activation of the hyperthermophilic Pf2001 esterase. Structure 2018, 26, 199–208.e3. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Rechavi, M.; Alibés, A.; Godzik, A. Contribution of electrostatic interactions, compactness and quaternary structure to protein thermostability: Lessons from structural genomics of Thermotoga maritima. J. Mol. Biol. 2006, 356, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Walden, H.; Bell, G.S.; Russell, R.J.M.; Siebers, B.; Hensel, R.; Taylor, G.L. Tiny TIM: A small, tetrameric, hyperthermostable triosephosphate isomerase. J. Mol. Biol. 2001, 306, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Skjold-Jørgensen, J.; Vind, J.; Moroz, O.V.; Blagova, E.; Bhatia, V.K.; Svendsen, A.; Wilson, K.S.; Bjerrum, M.J. Controlled lid-opening in Thermomyces lanuginosus lipase–An engineered switch for studying lipase function. Biochim. Biophys. Acta-Proteins Proteom. 2017, 1865, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, K.M.; Barbosa, L.R.S.; Lima, L.M.T.R.; Cortines, J.R.; Kalume, D.E.; Leal, I.C.R.; Mariz e Miranda, L.S.; De Souza, R.O.M.; Cordeiro, Y. Conformational dissection of Thermomyces lanuginosus lipase in solution. Biophys. Chem. 2014, 185, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.K.; Sørensen, T.R.; Kaspersen, J.D.; Silow, M.B.; Vind, J.; Pedersen, J.S.; Svendsen, A.; Otzen, D.E. Promoting protein self-association in non-glycosylated Thermomyces lanuginosus lipase based on crystal lattice contacts. Biochim. Biophys. Acta-Proteins Proteom. 2015, 1854, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Salameh, M.A.; Wiegel, J. Effects of detergents on activity, thermostability and aggregation of two alkalithermophilic lipases from Thermosyntropha lipolytica. Open Biochem. J. 2010, 4, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Lorente, G.; Palomo, J.M.; Fuentes, M.; Mateo, C.; Guisán, J.M.; Fernández-Lafuente, R. Self-assembly of Pseudomonas fluorescens lipase into bimolecular aggregates dramatically affects. Biotechnol. Bioeng. 2003, 82, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; Palomo, J.M.; Fernández-Lorente, G.; Illanes, A.; Guisán, J.M.; Fernández-Lafuente, R. Effect of lipase–lipase interactions in the activity, stability and specificity of a lipase from Alcaligenes sp. Enzyme Microb. Technol. 2006, 39, 259–264. [Google Scholar] [CrossRef]
- Otzen, D.E. Protein unfolding in detergents: Effect of micelle structure, ionic strength, pH, and temperature. Biophys. J. 2002, 83, 2219–2230. [Google Scholar] [CrossRef]
- Dünhaupt, A.; Lang, S.; Wagner, F. Pseudomonas cepacia lipase: Studies on aggregation, purification and on the cleavage of olive oil. Biotechnol. Lett. 1992, 14, 953–958. [Google Scholar] [CrossRef]
- Luisa Rúa, M.; Schmidt-Dannert, C.; Wahl, S.; Sprauer, A.; Schmid, R.D. Thermoalkalophilic lipase of Bacillus thermocatenulatus. Large-scale production, purification and properties: Aggregation behaviour and its effect on activity. J. Biotechnol. 1997, 56, 89–102. [Google Scholar] [CrossRef]
- Brunger, A.T. X-PLOR, version 3.1: A System for X-ray Crystallography and NMR; Yale University Press: New Haven, CT, USA, 1992. [Google Scholar]
- Jones, T.A.; Zou, J.Y.; Cowan, S.W.; Kjeldgaard, M. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 1991, 47, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, Y.; Fang, X.; Su, F.; Xu, L.; Yan, Y. Identification of a hot-spot to enhance: Candida rugosa lipase thermostability by rational design methods. RSC Adv. 2018, 8, 1948–1957. [Google Scholar] [CrossRef]
- Oguchi, Y.; Maeda, H.; Abe, K.; Nakajima, T.; Uchida, T.; Yamagata, Y. Hydrophobic interactions between the secondary structures on the molecular surface reinforce the alkaline stability of serine protease. Biotechnol. Lett. 2006, 28, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Gronenborn, A.M. Protein acrobatics in pairs–Dimerization via domain swapping. Curr. Opin. Struct. Biol. 2009, 19, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.J.; Choe, S.; Eisenberg, D. Domain swapping: Entangling alliances between proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 3127–3131. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Nishi, H.; Bryant, S.; Panchenko, A.R. Caught in self-interaction: Evolutionary and functional mechanisms of protein homooligomerization. Phys. Biol. 2011, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Panchenko, A.R. Mechanisms of protein oligomerization, the critical role of insertions and deletions in maintaining different oligomeric states. Proc. Natl. Acad. Sci. USA 2010, 107, 20352–20357. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Schymkowitz, J.W.H.; Wilkinson, H.R.; Itzhaki, L.S. Three-dimensional domain swapping in p13suc1 occurs in the unfolded state and is controlled by conserved proline residues. Proc. Natl. Acad. Sci. USA 2001, 98, 5596–5601. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Real Values | Coded Values 1 | ||||
|---|---|---|---|---|---|
| −1.267 | −1 | 0 | 1 | 1.267 | |
| pH | 5 | 5.4 | 40 | 8.6 | 9 |
| T (°C) | 30 | 32.1 | 7 | 47.9 | 50 |
| Run | Coded Values | Real Values | Residual Activity (%) 1 | |||||
|---|---|---|---|---|---|---|---|---|
| x1 | x2 | pH | T | mLip3 | dLip3 | |||
| Observed | Predicted | Observed | Predicted | |||||
| 1 | 1 | 1 | 8.6 | 47.9 | 1.5 | −0.09 | 40.20 | 46.08 |
| 2 | 1 | −1 | 8.6 | 32.1 | 48.3 | 47.10 | 82.50 | 81.24 |
| 3 | −1 | 1 | 5.4 | 47.9 | 83 | 84.83 | 83.80 | 88 |
| 4 | −1 | −1 | 5.4 | 32.1 | 84 | 86.22 | 84.30 | 81.36 |
| 5 | 1.267 | 0 | 9 | 40 | 2.2 | 4.51 | 54.90 | 51.74 |
| 6 | −1267 | 0 | 5 | 40 | 86.2 | 83.10 | 78.90 | 78.39 |
| 7 | 0 | 1.267 | 7 | 50 | 62.9 | 62.81 | 88.70 | 80.18 |
| 8 | 0 | −1.267 | 7 | 30 | 94.3 | 93.60 | 95.50 | 98.24 |
| 9 | 0 | 0 | 7 | 40 | 86.2 | 87.42 | 91.40 | 89.21 |
| 10 | 0 | 0 | 7 | 40 | 83.6 | 87.42 | 85.60 | 89.21 |
| 11 | 0 | 0 | 7 | 40 | 90.4 | 87.42 | 87.40 | 89.21 |
| 12 | 0 | 0 | 7 | 40 | 87.5 | 87.42 | 87.00 | 89.21 |
| 13 | 0 | 0 | 7 | 40 | 89.1 | 87.42 | 91.10 | 89.21 |
| Factor | SS 2 | DF 3 | MS 4 | F-Value | p-Value 5 |
|---|---|---|---|---|---|
| pH | 6935.61 | 1 | 6935.61 | 998.93 | 0.0000 |
| T | 1063.84 | 1 | 1063.84 | 153.23 | 0.0002 |
| pH2 | 3804.72 | 1 | 3804.72 | 547.99 | 0.0000 |
| pHT | 524.41 | 1 | 524.41 | 75.53 | 0.0010 |
| T2 | 169.87 | 1 | 169.87 | 24.47 | 0.0078 |
| Lack of fit | 27.74 | 3 | 9.25 | 1.33 | 0.3817 |
| Pure error | 27.77 | 4 | 6.94 | - | - |
| Total | 12,554.0 | 12 | - | - | - |
| Factor | SS 2 | DF 3 | MS 4 | F-Value | p-Value 5 |
|---|---|---|---|---|---|
| pH | 796.99 | 1 | 796.99 | 117.90 | 0.0004 |
| T | 366.61 | 1 | 366.61 | 54.23 | 0.0018 |
| pH2 | 1166.01 | 1 | 1166.01 | 172.49 | 0.0002 |
| pHT | 436.81 | 1 | 436.81 | 64.62 | 0.0013 |
| Lack of fit | 155.39 | 4 | 38.85 | 5.75 | 0.0594 |
| Pure error | 27.04 | 4 | 6.76 | - | - |
| Total | 2948.85 | 12 | - | - | - |
| mLip3 | dLip3 | |
|---|---|---|
| pH | 6.3 | 7.14 |
| Temperature (°C) | 35 | >50 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Cao, M.-E.; González, R.; Pernas, M.A.; Rúa, M.L. Contribution of the Oligomeric State to the Thermostability of Isoenzyme 3 from Candida rugosa. Microorganisms 2018, 6, 108. https://doi.org/10.3390/microorganisms6040108
Álvarez-Cao M-E, González R, Pernas MA, Rúa ML. Contribution of the Oligomeric State to the Thermostability of Isoenzyme 3 from Candida rugosa. Microorganisms. 2018; 6(4):108. https://doi.org/10.3390/microorganisms6040108
Chicago/Turabian StyleÁlvarez-Cao, María-Efigenia, Roberto González, María A. Pernas, and María Luisa Rúa. 2018. "Contribution of the Oligomeric State to the Thermostability of Isoenzyme 3 from Candida rugosa" Microorganisms 6, no. 4: 108. https://doi.org/10.3390/microorganisms6040108
APA StyleÁlvarez-Cao, M.-E., González, R., Pernas, M. A., & Rúa, M. L. (2018). Contribution of the Oligomeric State to the Thermostability of Isoenzyme 3 from Candida rugosa. Microorganisms, 6(4), 108. https://doi.org/10.3390/microorganisms6040108