
Immunoregulation in Fungal Diseases


Abstract
:1. Introduction
2. Immunoregulatory Cells
2.1. Dendritic Cells
2.2. Regulatory T Cells
3. Immunoregulatory Signaling Pathways
3.1. IL-10 Signaling
3.2. Programmed Cell Death Pathway
3.3. Cytotoxic T Lymphocyte-Associated Protein 4 Signaling
4. Unique Immunoregulatory Circumstances
4.1. Protective Tolerance
4.2. Immune Restoration Inflammatory Syndrome
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martins, N.; Ferreira, I.C.; Barros, L.; Silva, S.; Henriques, M. Candidiasis: Predisposing factors, prevention, diagnosis and alternative treatment. Mycopathologia 2014, 177, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Gonzalez, D.; Perusquia-Ortiz, A.M.; Hundeiker, M.; Bonifaz, A. Opportunistic yeast infections: Candidiasis, cryptococcosis, trichosporonosis and geotrichosis. J. Deutsch. Dermatol. Ges. 2013, 11, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Eggimann, P.; Garbino, J.; Pittet, D. Epidemiology of Candida species infections in critically ill non-immunosuppressed patients. Lancet Infect. Dis. 2003, 3, 685–702. [Google Scholar] [CrossRef]
- Rosen, L.B.; Freeman, A.F.; Yang, L.M.; Jutivorakool, K.; Olivier, K.N.; Angkasekwinai, N.; Suputtamongkol, Y.; Bennett, J.E.; Pyrgos, V.; Williamson, P.R.; et al. Anti-GM-CSF autoantibodies in patients with cryptococcal meningitis. J. Immunol. 2013, 190, 3959–3966. [Google Scholar] [CrossRef] [PubMed]
- Saijo, T.; Chen, J.; Chen, S.C.; Rosen, L.B.; Yi, J.; Sorrell, T.C.; Bennett, J.E.; Holland, S.M.; Browne, S.K.; Kwon-Chung, K.J. Anti-granulocyte-macrophage colony-stimulating factor autoantibodies are a risk factor for central nervous system infection by Cryptococcus gattii in otherwise immunocompetent patients. mBio 2014, 5, e00912–e00914. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Wuthrich, M.; Deepe, G.; Klein, B. Adaptive immunity to fungi. Cold Spring Harb. Perspect. Med. 2015, 5, a019612. [Google Scholar] [CrossRef] [PubMed]
- LeibundGut-Landmann, S.; Wuthrich, M.; Hohl, T.M. Immunity to fungi. Curr. Opin. Immunol. 2012, 24, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Herring, A.C.; Hernandez, Y.; Huffnagle, G.B.; Toews, G.B. Role and development of Th1/Th2 immune responses in the lungs. Semin. Respir. Crit. Care Med. 2004, 25, 3–10. [Google Scholar] [PubMed]
- Hernandez, Y.; Arora, S.; Erb-Downward, J.R.; McDonald, R.A.; Toews, G.B.; Huffnagle, G.B. Distinct roles for IL-4 and IL-10 in regulating T2 immunity during allergic bronchopulmonary mycosis. J. Immunol. 2005, 174, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.H.; Olszewski, M.A.; McDonald, R.A.; Wells, J.C.; Paine, R., 3rd; Huffnagle, G.B.; Toews, G.B. Role of granulocyte macrophage colony-stimulating factor in host defense against pulmonary cryptococcus neoformans infection during murine allergic bronchopulmonary mycosis. Am. J. Pathol. 2007, 170, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.H.; Osterholzer, J.J.; Choe, M.Y.; McDonald, R.A.; Olszewski, M.A.; Huffnagle, G.B.; Toews, G.B. Dual roles of CD40 on microbial containment and the development of immunopathology in response to persistent fungal infection in the lung. Am. J. Pathol. 2010, 177, 2459–2471. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Olszewski, M.A.; Tsang, T.M.; McDonald, R.A.; Toews, G.B.; Huffnagle, G.B. Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infect. Immun. 2011, 79, 1915–1926. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, A.; Asselin-Paturel, C.; Gilliet, M.; Crain, C.; Trinchieri, G.; Liu, Y.J.; O’Garra, A. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: Dependency on antigen dose and differential toll-like receptor ligation. J. Exp. Med. 2003, 197, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Cao, W.; Liu, Y.J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Gilfillan, S.; Vermi, W.; Wang, Y.; Colonna, M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral nk and CD8+ T cell accrual. Immunity 2010, 33, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yang, M.; Wang, Y.H.; Lande, R.; Gregorio, J.; Perng, O.A.; Qin, X.F.; Liu, Y.J.; Gilliet, M. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J. Exp. Med. 2007, 204, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Moseman, E.A.; Liang, X.; Dawson, A.J.; Panoskaltsis-Mortari, A.; Krieg, A.M.; Liu, Y.J.; Blazar, B.R.; Chen, W. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. J. Immunol. 2004, 173, 4433–4442. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Fukaya, T.; Eizumi, K.; Sato, Y.; Sato, K.; Shibazaki, A.; Otsuka, H.; Hijikata, A.; Watanabe, T.; Ohara, O.; et al. Plasmacytoid dendritic cells are crucial for the initiation of inflammation and T cell immunity in vivo. Immunity 2011, 35, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Ochando, J.C.; Homma, C.; Yang, Y.; Hidalgo, A.; Garin, A.; Tacke, F.; Angeli, V.; Li, Y.; Boros, P.; Ding, Y.; et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat. Immunol. 2006, 7, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Ortiz, Z.G.; Specht, C.A.; Wang, J.P.; Lee, C.K.; Bartholomeu, D.C.; Gazzinelli, R.T.; Levitz, S.M. Toll-like receptor 9-dependent immune activation by unmethylated CPG motifs in aspergillus fumigatus DNA. Infect. Immun. 2008, 76, 2123–2129. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Ortiz, Z.G.; Lee, C.K.; Wang, J.P.; Boon, L.; Specht, C.A.; Levitz, S.M. A nonredundant role for plasmacytoid dendritic cells in host defense against the human fungal pathogen aspergillus fumigatus. Cell Host Microbe 2011, 9, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Liu, Y.J. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J. Exp. Med. 2002, 195, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Belz, G.T.; Behrens, G.M.; Smith, C.M.; Miller, J.F.; Jones, C.; Lejon, K.; Fathman, C.G.; Mueller, S.N.; Shortman, K.; Carbone, F.R.; et al. The CD8α+ dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J. Exp. Med. 2002, 196, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Dudziak, D.; Heidkamp, G.F.; Fiorese, C.; Bonito, A.J.; Inaba, K.; Nussenzweig, M.C.; Steinman, R.M. CD8+CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J. Immunol. 2008, 181, 6923–6933. [Google Scholar] [CrossRef] [PubMed]
- Min, S.Y.; Park, K.S.; Cho, M.L.; Kang, J.W.; Cho, Y.G.; Hwang, S.Y.; Park, M.J.; Yoon, C.H.; Min, J.K.; Lee, S.H.; et al. Antigen-induced, tolerogenic CD11c+,CD11b+ dendritic cells are abundant in Peyer’s patches during the induction of oral tolerance to type II collagen and suppress experimental collagen-induced arthritis. Arthritis Rheumatol. 2006, 54, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, M.A.; Rathinam, C.; Flavell, R.A. Pancreatic islet expression of chemokine CCL2 suppresses autoimmune diabetes via tolerogenic CD11c+ CD11b+ dendritic cells. Proc. Natl. Acad. Sci. USA 2012, 109, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.H. In vivo function of langerhans cells and dermal dendritic cells. Trends Immunol. 2010, 31, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Igyarto, B.Z.; Haley, K.; Ortner, D.; Bobr, A.; Gerami-Nejad, M.; Edelson, B.T.; Zurawski, S.M.; Malissen, B.; Zurawski, G.; Berman, J.; et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific t helper cell responses. Immunity 2011, 35, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Igyarto, B.Z.; Gerami-Nejad, M.; Kumamoto, Y.; Mohammed, J.; Jarrett, E.; Drummond, R.A.; Zurawski, S.M.; Zurawski, G.; Berman, J.; et al. Candida albicans morphology and dendritic cell subsets determine t helper cell differentiation. Immunity 2015, 42, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Osterholzer, J.J.; Chen, G.H.; Olszewski, M.A.; Curtis, J.L.; Huffnagle, G.B.; Toews, G.B. Accumulation of CD11b+ lung dendritic cells in response to fungal infection results from the CCR2-mediated recruitment and differentiation of Ly-6Chigh Monocytes. J. Immunol. 2009, 183, 8044–8053. [Google Scholar] [CrossRef] [PubMed]
- Osterholzer, J.J.; Curtis, J.L.; Polak, T.; Ames, T.; Chen, G.H.; McDonald, R.; Huffnagle, G.B.; Toews, G.B. CCR2 mediates conventional dendritic cell recruitment and the formation of bronchovascular mononuclear cell infiltrates in the lungs of mice infected with Cryptococcus neoformans. J. Immunol. 2008, 181, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Osterholzer, J.J.; Chen, G.H.; Olszewski, M.A.; Zhang, Y.M.; Curtis, J.L.; Huffnagle, G.B.; Toews, G.B. Chemokine receptor 2-mediated accumulation of fungicidal exudate macrophages in mice that clear cryptococcal lung infection. Am. J. Pathol. 2011, 178, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Traynor, T.R.; Kuziel, W.A.; Toews, G.B.; Huffnagle, G.B. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J. Immunol. 2000, 164, 2021–2027. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, W.A.; Deepe, G.S., Jr. The CCL7-CCL2-CCR2 axis regulates IL-4 production in lungs and fungal immunity. J. Immunol. 2009, 183, 1964–1974. [Google Scholar]
- Hohl, T.M.; Rivera, A.; Lipuma, L.; Gallegos, A.; Shi, C.; Mack, M.; Pamer, E.G. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe 2009, 6, 470–481. [Google Scholar] [PubMed]
- Vecchiarelli, A.; Retini, C.; Monari, C.; Tascini, C.; Bistoni, F.; Kozel, T.R. Purified capsular polysaccharide of Cryptococcus neoformans induces interleukin-10 secretion by human monocytes. Infect. Immun. 1996, 64, 2846–2849. [Google Scholar] [PubMed]
- Vecchiarelli, A.; Pietrella, D.; Lupo, P.; Bistoni, F.; McFadden, D.C.; Casadevall, A. The polysaccharide capsule of Cryptococcus neoformans interferes with human dendritic cell maturation and activation. J. Leukoc. Biol. 2003, 74, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Monari, C.; Baldelli, F.; Pietrella, D.; Retini, C.; Tascini, C.; Francisci, D.; Bistoni, F.; Vecchiarelli, A. Monocyte dysfunction in patients with acquired immunodeficiency syndrome (AIDS) versus Cryptococcus neoformans. J. Infect. 1997, 35, 257–263. [Google Scholar] [CrossRef]
- Murdock, B.J.; Teitz-Tennenbaum, S.; Chen, G.H.; Dils, A.J.; Malachowski, A.N.; Curtis, J.L.; Olszewski, M.A.; Osterholzer, J.J. Early or late IL-10 blockade enhances Th1 and Th17 effector responses and promotes fungal clearance in mice with cryptococcal lung infection. J. Immunol. 2014, 193, 4107–4116. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M. Regulatory/suppressor T cells in health and disease. Arthritis Rheumatol. 2004, 50, 2721–2724. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.; Powrie, F. Regulatory T cells. Curr. Opin. Pharmacol. 2004, 4, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Sakaguchi, S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 2010, 11, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Sakaguchi, S. Regulatory T cells in immune surveillance and treatment of cancer. Semin. Cancer Biol. 2006, 16, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Tarbell, K. Regulatory T cells in the control of host-microorganism interactions*. Annu. Rev. Immunol. 2009, 27, 551–589. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.J.; Powrie, F. Regulatory T cells reinforce intestinal homeostasis. Immunity 2009, 31, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.J.; Sakaguchi, S. Regulatory T cells in transplantation tolerance. Nat. Rev. Immunol. 2003, 3, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Maggi, E.; Cosmi, L.; Liotta, F.; Romagnani, P.; Romagnani, S.; Annunziato, F. Thymic regulatory T cells. Autoimmun. Rev. 2005, 4, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Lan, R.Y.; Ansari, A.A.; Lian, Z.X.; Gershwin, M.E. Regulatory T cells: Development, function and role in autoimmunity. Autoimmun. Rev. 2005, 4, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.H. Natural regulatory T cells and self-tolerance. Nat. Immunol. 2005, 6, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kitani, A.; Fuss, I.; Pedersen, A.; Harada, N.; Nawata, H.; Strober, W. TGF-β 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J. Immunol. 2004, 172, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Roncarolo, M.G.; Gregori, S.; Battaglia, M.; Bacchetta, R.; Fleischhauer, K.; Levings, M.K. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol. Rev. 2006, 212, 28–50. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y. Regulatory T cells and infection: A dangerous necessity. Nat. Rev. Immunol. 2007, 7, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Von Boehmer, H. Mechanisms of suppression by suppressor T cells. Nat. Immunol. 2005, 6, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Lukas, D.; Clausen, B.E.; Yogev, N. Dendritic cells as gatekeepers of tolerance. Semin. Immunopathol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Conti, H.R.; Zheng, L.; Peterson, A.C.; Mathern, D.R.; Hernandez-Santos, N.; Edgerton, M.; Gaffen, S.L.; Lenardo, M.J. CD4+CD25+Foxp3+ regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse candida albicans Th17 cell infection model. Immunity 2011, 34, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Sutmuller, R.; Hermann, C.; van der Graaf, C.A.; van der Meer, J.W.; van Krieken, J.H.; Hartung, T.; Adema, G.; Kullberg, B.J. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J. Immunol. 2004, 172, 3712–3718. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.P.; Cavassani, K.A.; Massafera Tristao, F.S.; Campanelli, A.P.; Martinez, R.; Rossi, M.A.; Silva, J.S. CCR5-dependent regulatory T cell migration mediates fungal survival and severe immunosuppression. J. Immunol. 2008, 180, 3049–3056. [Google Scholar] [CrossRef] [PubMed]
- Schulze, B.; Piehler, D.; Eschke, M.; von Buttlar, H.; Kohler, G.; Sparwasser, T.; Alber, G. CD4+Foxp3+ regulatory T cells suppress fatal T helper 2 cell immunity during pulmonary fungal infection. Eur. J. Immunol. 2014, 44, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, D.L.; Smith, K.D.; Kotov, D.I.; Nielsen, J.N.; Bohjanen, P.R.; Nielsen, K. Regulatory T cell induction and retention in the lungs drives suppression of detrimental type 2 Th cells during pulmonary cryptococcal infection. J. Immunol. 2016, 196, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Kroetz, D.N.; Deepe, G.S., Jr. CCR5 dictates the equilibrium of proinflammatory IL-17+ and regulatory Foxp3+ T cells in fungal infection. J. Immunol. 2010, 184, 5224–5231. [Google Scholar] [CrossRef] [PubMed]
- Montagnoli, C.; Bozza, S.; Gaziano, R.; Zelante, T.; Bonifazi, P.; Moretti, S.; Bellocchio, S.; Pitzurra, L.; Romani, L. Immunity and tolerance to aspergillus fumigatus. Novartis Found. Symp. 2006, 279, 66–77. [Google Scholar] [PubMed]
- Whibley, N.; Maccallum, D.M.; Vickers, M.A.; Zafreen, S.; Waldmann, H.; Hori, S.; Gaffen, S.L.; Gow, N.A.; Barker, R.N.; Hall, A.M. Expansion of Foxp3+ T-cell populations by Candida albicans enhances both Th17-cell responses and fungal dissemination after intravenous challenge. Eur. J. Immunol. 2014, 44, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Loures, F.V.; Pina, A.; Felonato, M.; Calich, V.L. TLR2 is a negative regulator of Th17 cells and tissue pathology in a pulmonary model of fungal infection. J. Immunol. 2009, 183, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.C.; de Oliveira, R.T.; da Silva, R.M.; Blotta, M.H.; Mamoni, R.L. Involvement of regulatory T cells in the immunosuppression characteristic of patients with paracoccidioidomycosis. Infect. Immun. 2010, 78, 4392–4401. [Google Scholar] [CrossRef] [PubMed]
- Bonifazi, P.; Zelante, T.; D’Angelo, C.; de Luca, A.; Moretti, S.; Bozza, S.; Perruccio, K.; Iannitti, R.G.; Giovannini, G.; Volpi, C.; et al. Balancing inflammation and tolerance in vivo through dendritic cells by the commensal Candida albicans. Mucosal Immunol. 2009, 2, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Whibley, N.; Gaffen, S.L. Brothers in arms: Th17 and treg responses in Candida albicans immunity. PLoS Pathog. 2014, 10, e1004456. [Google Scholar] [CrossRef] [PubMed]
- Sitrin, J.; Ring, A.; Garcia, K.C.; Benoist, C.; Mathis, D. Regulatory T cells control NK cells in an insulitic lesion by depriving them of IL-2. J. Exp. Med. 2013, 210, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Montagnoli, C.; Zelante, T.; Bonifazi, P.; Bozza, S.; Moretti, S.; D’Angelo, C.; Vacca, C.; Boon, L.; Bistoni, F.; et al. Functional yet balanced reactivity to Candida albicans requires TRIF, MyD88, and IDO-dependent inhibition of Rorc. J. Immunol. 2007, 179, 5999–6008. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Ouyang, W. Regulation of interleukin-10 expression. Adv. Exp. Med. Biol. 2016, 941, 89–116. [Google Scholar] [PubMed]
- Akbari, O.; DeKruyff, R.H.; Umetsu, D.T. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat. Immunol. 2001, 2, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, D.F.; Bond, M.W.; Mosmann, T.R. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. 1989, 170, 2081–2095. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, N.; Schenten, D.; Nish, S.A.; Walther, Z.; Gagliani, N.; Flavell, R.A.; Reizis, B.; Shen, Z.; Fox, J.G.; Iwasaki, A.; et al. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nat. Commun. 2012, 3, 1120. [Google Scholar] [CrossRef] [PubMed]
- Freitas do Rosario, A.P.; Lamb, T.; Spence, P.; Stephens, R.; Lang, A.; Roers, A.; Muller, W.; O’Garra, A.; Langhorne, J. IL-27 promotes IL-10 production by effector Th1 CD4+ T cells: A critical mechanism for protection from severe immunopathology during malaria infection. J. Immunol. 2012, 188, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.; Lohler, J.; Rennick, D.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Cyktor, J.C.; Turner, J. Interleukin-10 and immunity against prokaryotic and eukaryotic intracellular pathogens. Infect. Immun. 2011, 79, 2964–2973. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Lortholary, O.; Improvisi, L.; Rayhane, N.; Gray, F.; Fitting, C.; Cavaillon, J.M.; Dromer, F. Cytokine profiles of AIDS patients are similar to those of mice with disseminated Cryptococcus neoformans infection. Infect. Immun. 1999, 67, 6314–6320. [Google Scholar] [PubMed]
- Blackstock, R.; Buchanan, K.L.; Adesina, A.M.; Murphy, J.W. Differential regulation of immune responses by highly and weakly virulent Cryptococcus neoformans isolates. Infect. Immun. 1999, 67, 3601–3609. [Google Scholar] [PubMed]
- Sun, R.T.; Tian, W.J.; Xing, X.W.; Gao, S.H.; Wang, S.B. Association of cytokine gene polymorphisms with susceptibility to invasive candidiasis. Genet. Mol. Res. 2015, 14, 6859–6864. [Google Scholar] [CrossRef] [PubMed]
- Sainz, J.; Hassan, L.; Perez, E.; Romero, A.; Moratalla, A.; Lopez-Fernandez, E.; Oyonarte, S.; Jurado, M. Interleukin-10 promoter polymorphism as risk factor to develop invasive pulmonary aspergillosis. Immunol. Lett. 2007, 109, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Torres, A.; Jones-Carson, J.; Wagner, R.D.; Warner, T.; Balish, E. Early resistance of interleukin-10 knockout mice to acute systemic candidiasis. Infect. Immun. 1999, 67, 670–674. [Google Scholar] [PubMed]
- Sahaza, J.H.; Suarez-Alvarez, R.; Estrada-Barcenas, D.A.; Perez-Torres, A.; Taylor, M.L. Profile of cytokines in the lungs of BALB/c mice after intra-nasal infection with Histoplasma capsulatum mycelial propagules. Comp. Immunol. Microbiol. Infect. Dis. 2015, 41, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mirkov, I.; Demenesku, J.; Popov Aleksandrov, A.; Ninkov, M.; Glamoclija, J.; Kataranovski, D.; Kataranovski, M. Strain differences in the immune mechanisms of resistance of immunocompetent rats to pulmonary aspergillosis. Immunobiology 2015, 220, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [PubMed]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.S.; Zhang, C.; Lee, C.H. Myeloid-derived suppressor cells impair alveolar macrophages through PD-1 receptor ligation during pneumocystis pneumonia. Infect. Immun. 2015, 83, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K.; et al. Expression of programmed death 1 ligands by murine T cells and apc. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an itim motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, T.L.; Thomson, A.W. The status of PD-L1 (B7-H1) on tolerogenic APCs. Eur. J. Immunol. 2011, 41, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Terawaki, S.; Honjo, T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int. Immunol. 2005, 17, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G.; et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005, 65, 1089–1096. [Google Scholar] [PubMed]
- Okudaira, K.; Hokari, R.; Tsuzuki, Y.; Okada, Y.; Komoto, S.; Watanabe, C.; Kurihara, C.; Kawaguchi, A.; Nagao, S.; Azuma, M.; et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int. J. Oncol. 2009, 35, 741–749. [Google Scholar] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, M.; Pareek, V.; Cheng, H.; Narasimhulu, D.M.; Zang, X. Immune checkpoint blockade in human cancer therapy: Lung cancer and hematologic malignancies. Immunotherapy 2016, 8, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Janik, J.E.; Abu-Eid, R.; Mkrtichyan, M.; Khleif, S.N. Programmed death-1 & its ligands: Promising targets for cancer immunotherapy. Immunotherapy 2015, 7, 777–792. [Google Scholar] [PubMed]
- Kaufmann, D.E.; Walker, B.D. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J. Immunol. 2009, 182, 5891–5897. [Google Scholar] [CrossRef] [PubMed]
- Eichbaum, Q. PD-1 signaling in HIV and chronic viral infection—Potential for therapeutic intervention? Curr. Med. Chem. 2011, 18, 3971–3980. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Mayer-Barber, K.D.; Feng, C.G.; Sharpe, A.H.; Sher, A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J. Immunol. 2011, 186, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.H.; Johanns, T.M.; Ertelt, J.M.; Way, S.S. PDL-1 blockade impedes T cell expansion and protective immunity primed by attenuated listeria monocytogenes. J. Immunol. 2008, 180, 7553–7557. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, R.; Gigley, J.P.; Weiss, L.M.; Khan, I.A. Control of toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1-PDL-1 blockade. Proc. Natl. Acad. Sci. USA 2011, 108, 9196–9201. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Jain, N.; Wang, X.; Fries, B.C. Cryptococcus neoformans variants generated by phenotypic switching differ in virulence through effects on macrophage activation. Infect. Immun. 2010, 78, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Lazar-Molnar, E.; Gacser, A.; Freeman, G.J.; Almo, S.C.; Nathenson, S.G.; Nosanchuk, J.D. The PD-1/PD-L costimulatory pathway critically affects host resistance to the pathogenic fungus histoplasma capsulatum. Proc. Natl. Acad. Sci. USA 2008, 105, 2658–2663. [Google Scholar] [CrossRef] [PubMed]
- Cacere, C.R.; Mendes-Giannini, M.J.; Fontes, C.J.; Kono, A.; Duarte, A.J.; Benard, G. Altered expression of the costimulatory molecules CD80, CD86, CD152, PD-1 and ICOS on T-cells from paracoccidioidomycosis patients: Lack of correlation with T-cell hyporesponsiveness. Clin. Immunol. 2008, 129, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, H.; Muskens, F.; Willart, M.; Hijdra, D.; van Assema, F.B.; Coyle, A.J.; Hoogsteden, H.C.; Lambrecht, B.N. Contribution of the PD-1 ligands/PD-1 signaling pathway to dendritic cell-mediated CD4+ T cell activation. Eur. J. Immunol. 2006, 36, 2472–2482. [Google Scholar] [CrossRef] [PubMed]
- Spec, A.; Shindo, Y.; Burnham, C.A.; Wilson, S.; Ablordeppey, E.A.; Beiter, E.R.; Chang, K.; Drewry, A.M.; Hotchkiss, R.S. T cells from patients with Candida sepsis display a suppressive immunophenotype. Crit. Care 2016, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Burnham, C.A.; Compton, S.M.; Rasche, D.P.; Mazuski, R.J.; McDonough, J.S.; Unsinger, J.; Korman, A.J.; Green, J.M.; Hotchkiss, R.S. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit. Care 2013, 17, R85. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Rathmell, J.C.; Gray, G.S.; Thompson, C.B.; Leiden, J.M.; Alegre, M.L. Cytotoxic T lymphocyte antigen 4 (CTLA4) blockade accelerates the acute rejection of cardiac allografts in CD28-deficient mice: CTLA4 can function independently of CD28. J. Exp. Med. 1998, 188, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Azuma, M.; Ito, D.; Yagita, H.; Okumura, K.; Phillips, J.H.; Lanier, L.L.; Somoza, C. B70 antigen is a second ligand for CTLA-4 and CD28. Nature 1993, 366, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J.; Su, G.H.; Zuckerman, L.A.; Nabavi, N.; Jellis, C.L.; Gray, G.S.; Miller, J.; Bluestone, J.A. Expression and functional significance of an additional ligand for CTLA-4. Proc. Natl. Acad. Sci. USA 1993, 90, 11054–11058. [Google Scholar] [CrossRef] [PubMed]
- Oosterwegel, M.A.; Greenwald, R.J.; Mandelbrot, D.A.; Lorsbach, R.B.; Sharpe, A.H. CTLA-4 and T cell activation. Curr. Opin. Immunol. 1999, 11, 294–300. [Google Scholar] [CrossRef]
- Reiser, H.; Freeman, G.J.; Razi-Wolf, Z.; Gimmi, C.D.; Benacerraf, B.; Nadler, L.M. Murine B7 antigen provides an efficient costimulatory signal for activation of murine T lymphocytes via the T-cell receptor/CD3 complex. Proc. Natl. Acad. Sci. USA 1992, 89, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Gimmi, C.D.; Freeman, G.J.; Gribben, J.G.; Sugita, K.; Freedman, A.S.; Morimoto, C.; Nadler, L.M. B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc. Natl. Acad. Sci. USA 1991, 88, 6575–6579. [Google Scholar] [CrossRef] [PubMed]
- Kearney, E.R.; Walunas, T.L.; Karr, R.W.; Morton, P.A.; Loh, D.Y.; Bluestone, J.A.; Jenkins, M.K. Antigen-dependent clonal expansion of a trace population of antigen-specific CD4+ T cells in vivo is dependent on CD28 costimulation and inhibited by CTLA-4. J. Immunol. 1995, 155, 1032–1036. [Google Scholar] [PubMed]
- Freeman, G.J.; Borriello, F.; Hodes, R.J.; Reiser, H.; Hathcock, K.S.; Laszlo, G.; McKnight, A.J.; Kim, J.; Du, L.; Lombard, D.B.; et al. Uncovering of functional alternative CTLA-4 counter-receptor in B7-deficient mice. Science 1993, 262, 907–909. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Wallace, P.M.; Johnson, J.; Gibson, M.G.; Greene, J.L.; Ledbetter, J.A.; Singh, C.; Tepper, M.A. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science 1992, 257, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Bakker, C.Y.; Bluestone, J.A. CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 1996, 183, 2541–2550. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Probst, H.C.; McCoy, K.; Okazaki, T.; Honjo, T.; van den Broek, M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat. Immunol. 2005, 6, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Campanelli, A.P.; Martins, G.A.; Souto, J.T.; Pereira, M.S.; Livonesi, M.C.; Martinez, R.; Silva, J.S. Fas-fas ligand (CD95-CD95L) and cytotoxic T lymphocyte antigen-4 engagement mediate T cell unresponsiveness in patients with paracoccidioidomycosis. J. Infect. Dis. 2003, 187, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Pietrella, D.; Perito, S.; Bistoni, F.; Vecchiarelli, A. Cytotoxic t lymphocyte antigen costimulation influences T-cell activation in response to Cryptococcus neoformans. Infect. Immun. 2001, 69, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- McGaha, T.; Murphy, J.W. CTLA-4 down-regulates the protective anticryptococcal cell-mediated immune response. Infect. Immun. 2000, 68, 4624–4630. [Google Scholar] [CrossRef] [PubMed]
- Krummey, S.M.; Floyd, T.L.; Liu, D.; Wagener, M.E.; Song, M.; Ford, M.L. Candida-elicited murine Th17 cells express high CTLA-4 compared with Th1 cells and are resistant to costimulation blockade. J. Immunol. 2014, 192, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Romani, L.; Puccetti, P. Protective tolerance to fungi: The role of IL-10 and tryptophan catabolism. Trends Microbiol. 2006, 14, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Montagnoli, C.; Bacci, A.; Bozza, S.; Gaziano, R.; Mosci, P.; Sharpe, A.H.; Romani, L. B7/CD28-dependent CD4+CD25+ regulatory T cells are essential components of the memory-protective immunity to Candida albicans. J. Immunol. 2002, 169, 6298–6308. [Google Scholar] [CrossRef] [PubMed]
- Montagnoli, C.; Fallarino, F.; Gaziano, R.; Bozza, S.; Bellocchio, S.; Zelante, T.; Kurup, W.P.; Pitzurra, L.; Puccetti, P.; Romani, L. Immunity and tolerance to aspergillus involve functionally distinct regulatory T cells and tryptophan catabolism. J. Immunol. 2006, 176, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Romani, L.; Bistoni, F.; Perruccio, K.; Montagnoli, C.; Gaziano, R.; Bozza, S.; Bonifazi, P.; Bistoni, G.; Rasi, G.; Velardi, A.; et al. Thymosin α1 activates dendritic cell tryptophan catabolism and establishes a regulatory environment for balance of inflammation and tolerance. Blood 2006, 108, 2265–2274. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Volpi, C.; Fallarino, F.; Bozza, S.; Bianchi, R.; Vacca, C.; Orabona, C.; Belladonna, M.L.; Ayroldi, E.; Nocentini, G.; et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat. Med. 2007, 13, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Romani, L.; Puccetti, P. Immune regulation and tolerance to fungi in the lungs and skin. Chem. Immunol. Allergy 2008, 94, 124–137. [Google Scholar] [PubMed]
- Shelburne, S.A., 3rd; Hamill, R.J.; Rodriguez-Barradas, M.C.; Greenberg, S.B.; Atmar, R.L.; Musher, D.W.; Gathe, J.C., Jr.; Visnegarwala, F.; Trautner, B.W. Immune reconstitution inflammatory syndrome: Emergence of a unique syndrome during highly active antiretroviral therapy. Medicine 2002, 81, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, D.M.; Venter, W.D.; Feldman, C.; Van Rie, A. Incidence and risk factors for the immune reconstitution inflammatory syndrome in HIV patients in South Africa: A prospective study. AIDS 2008, 22, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Shelburne, S.A.; Visnegarwala, F.; Darcourt, J.; Graviss, E.A.; Giordano, T.P.; White, A.C., Jr.; Hamill, R.J. Incidence and risk factors for immune reconstitution inflammatory syndrome during highly active antiretroviral therapy. AIDS 2005, 19, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Boulougoura, A.; Sereti, I. HIV infection and immune activation: The role of coinfections. Curr. Opin. HIV AIDS 2016, 11, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Jenny-Avital, E.R.; Abadi, M. Immune reconstitution cryptococcosis after initiation of successful highly active antiretroviral therapy. Clin. Infect. Dis. 2002, 35, e128–e133. [Google Scholar] [CrossRef] [PubMed]
- French, M.A.; Mallal, S.A.; Dawkins, R.L. Zidovudine-induced restoration of cell-mediated immunity to mycobacteria in immunodeficient HIV-infected patients. AIDS 1992, 6, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Woods, M.L., 2nd; MacGinley, R.; Eisen, D.P.; Allworth, A.M. HIV combination therapy: Partial immune restitution unmasking latent cryptococcal infection. AIDS 1998, 12, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- French, M.A. HIV/AIDS: Immune reconstitution inflammatory syndrome: A reappraisal. Clin. Infect. Dis. 2009, 48, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Haddow, L.J.; Colebunders, R.; Meintjes, G.; Lawn, S.D.; Elliott, J.H.; Manabe, Y.C.; Bohjanen, P.R.; Sungkanuparph, S.; Easterbrook, P.J.; French, M.A.; et al. Cryptococcal immune reconstitution inflammatory syndrome in HIV-1-infected individuals: Proposed clinical case definitions. Lancet Infect. Dis. 2010, 10, 791–802. [Google Scholar] [CrossRef]
- Maziarz, E.K.; Perfect, J.R. Cryptococcosis. Infect. Dis. Clin. North Am. 2016, 30, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, L.R.; Mahnke, Y.; Hodge, J.N.; Porter, B.O.; Barber, D.L.; DerSimonian, R.; Greenwald, J.H.; Roby, G.; Mican, J.; Sher, A.; et al. Elevated frequencies of highly activated CD4+ T cells in HIV+ patients developing immune reconstitution inflammatory syndrome. Blood 2010, 116, 3818–3827. [Google Scholar] [CrossRef] [PubMed]
- Sereti, I.; Rodger, A.J.; French, M.A. Biomarkers in immune reconstitution inflammatory syndrome: Signals from pathogenesis. Curr. Opin. HIV AIDS 2010, 5, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.B.; Yong, Y.K.; Tan, H.Y.; Kamarulzaman, A.; Tan, L.H.; Lim, A.; James, I.; French, M.; Price, P. Immunological profiles of immune restoration disease presenting as mycobacterial lymphadenitis and cryptococcal meningitis. HIV Med. 2008, 9, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Meya, D.B.; Okurut, S.; Zziwa, G.; Rolfes, M.A.; Kelsey, M.; Cose, S.; Joloba, M.; Naluyima, P.; Palmer, B.E.; Kambugu, A.; et al. Cellular immune activation in cerebrospinal fluid from ugandans with cryptococcal meningitis and immune reconstitution inflammatory syndrome. J. Infect. Dis. 2015, 211, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Boulware, D.R.; Bonham, S.C.; Meya, D.B.; Wiesner, D.L.; Park, G.S.; Kambugu, A.; Janoff, E.N.; Bohjanen, P.R. Paucity of initial cerebrospinal fluid inflammation in cryptococcal meningitis is associated with subsequent immune reconstitution inflammatory syndrome. J. Infect. Dis. 2010, 202, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Lortholary, O.; Alexander, B.D.; Gupta, K.L.; John, G.T.; Pursell, K.; Munoz, P.; Klintmalm, G.B.; Stosor, V.; del Busto, R.; et al. An immune reconstitution syndrome-like illness associated with Cryptococcus neoformans infection in organ transplant recipients. Clin. Infect. Dis. 2005, 40, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Y.; Munoz, P.; Torre-Cisneros, J.; Aguado, J.M.; Lattes, R.; Montejo, M.; Garcia-Reyne, A.; Bouza, E.; Valerio, M.; Lara, R.; et al. Mycobacterium tuberculosis-associated immune reconstitution syndrome in solid-organ transplant recipients. Transplantation 2013, 95, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, N.; Reyes, J.; Kusne, S.; Martin, M.; Fung, J. Cryptococcal meningitis after liver transplantation. Transplantation 1996, 61, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Mihu, M.R.; Pattabhi, R.; Nosanchuk, J.D. The impact of antifungals on toll-like receptors. Front. Microbiol. 2014, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- McLin, V.A.; Belli, D.C.; Posfay-Barbe, K.M. Immune reconstitution inflammatory syndrome and solid organ transplant recipients: Are children protected? Pediatr. Transplant. 2010, 14, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Sirinavin, S.; Intusoma, U.; Tuntirungsee, S. Mother-to-child transmission of Cryptococcus neoformans. Pediatr. Infect. Dis. J. 2004, 23, 278–279. [Google Scholar] [CrossRef] [PubMed]
- Einsiedel, L.; Gordon, D.L.; Dyer, J.R. Paradoxical inflammatory reaction during treatment of cryptococcus neoformans var. Gattii meningitis in an HIV-seronegative woman. Clin. Infect. Dis. 2004, 39, e78–e82. [Google Scholar] [CrossRef] [PubMed]
- Annapureddy, S.R.; Masterson, S.W.; David, H.G.; Greig, J.R. Post partum osteomyelitis due to Cryptococcus neoformans. Scand. J. Infect. Dis. 2007, 39, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Hamer, L.; Castillo, J. Coccidioidomycosis presenting as a massive pleural effusion in a postpartum woman. Indian J. Chest Dis. Allied Sci. 2006, 48, 59–62. [Google Scholar] [PubMed]
- Spinello, I.M.; Johnson, R.H.; Baqi, S. Coccidioidomycosis and pregnancy: A review. Ann. N. Y. Acad. Sci. 2007, 1111, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Poole, J.A.; Claman, H.N. Immunology of pregnancy. Implications for the mother. Clin. Rev. Allergy Immunol. 2004, 26, 161–170. [Google Scholar] [CrossRef]
- Koguchi, Y.; Kawakami, K. Cryptococcal infection and Th1-Th2 cytokine balance. Int. Rev. Immunol. 2002, 21, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Decken, K.; Kohler, G.; Palmer-Lehmann, K.; Wunderlin, A.; Mattner, F.; Magram, J.; Gately, M.K.; Alber, G. Interleukin-12 is essential for a protective Th1 response in mice infected with Cryptococcus neoformans. Infect. Immun. 1998, 66, 4994–5000. [Google Scholar] [PubMed]
- Elenkov, I.J.; Wilder, R.L.; Bakalov, V.K.; Link, A.A.; Dimitrov, M.A.; Fisher, S.; Crane, M.; Kanik, K.S.; Chrousos, G.P. IL-12, TNF-α, and hormonal changes during late pregnancy and early postpartum: Implications for autoimmune disease activity during these times. J. Clin. Endocrinol. Metab. 2001, 86, 4933–4938. [Google Scholar] [PubMed]
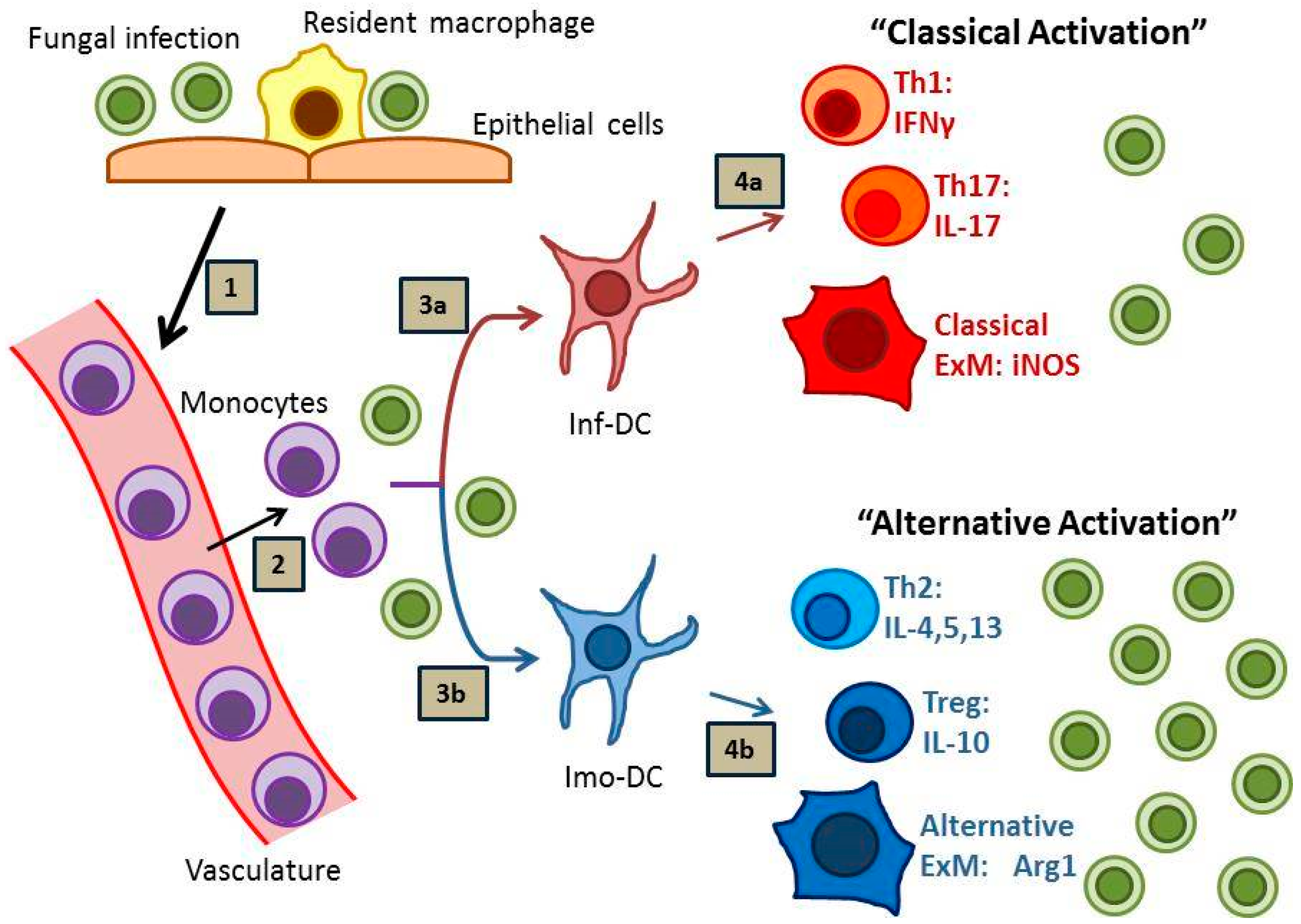

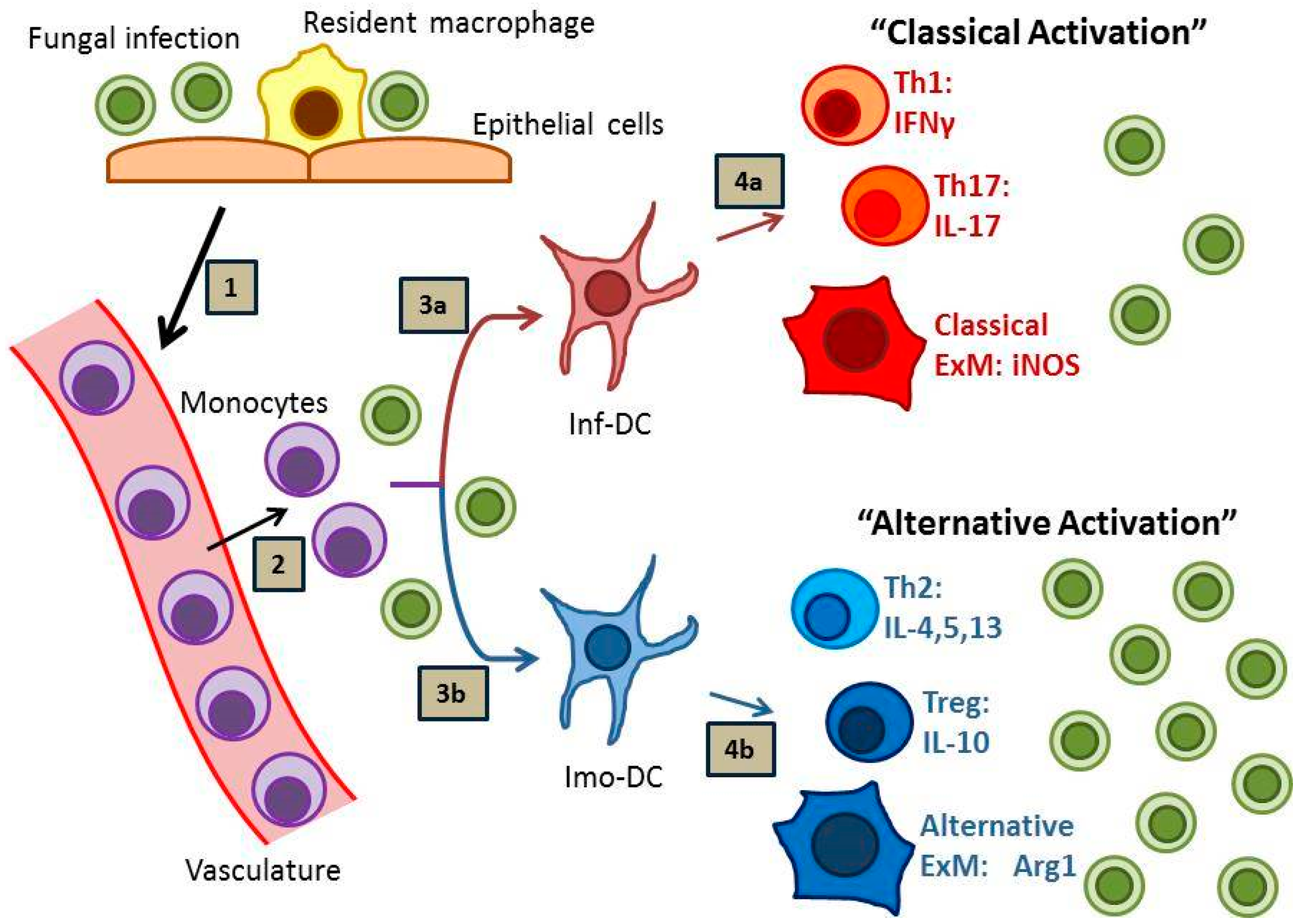{kind=link}
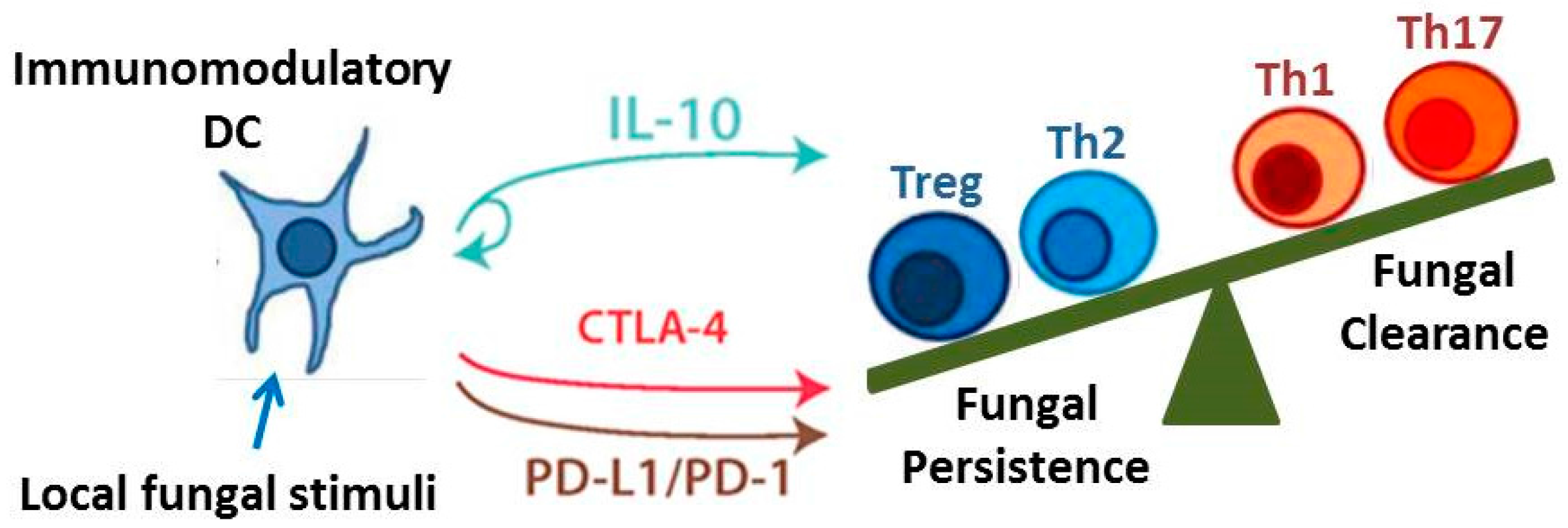
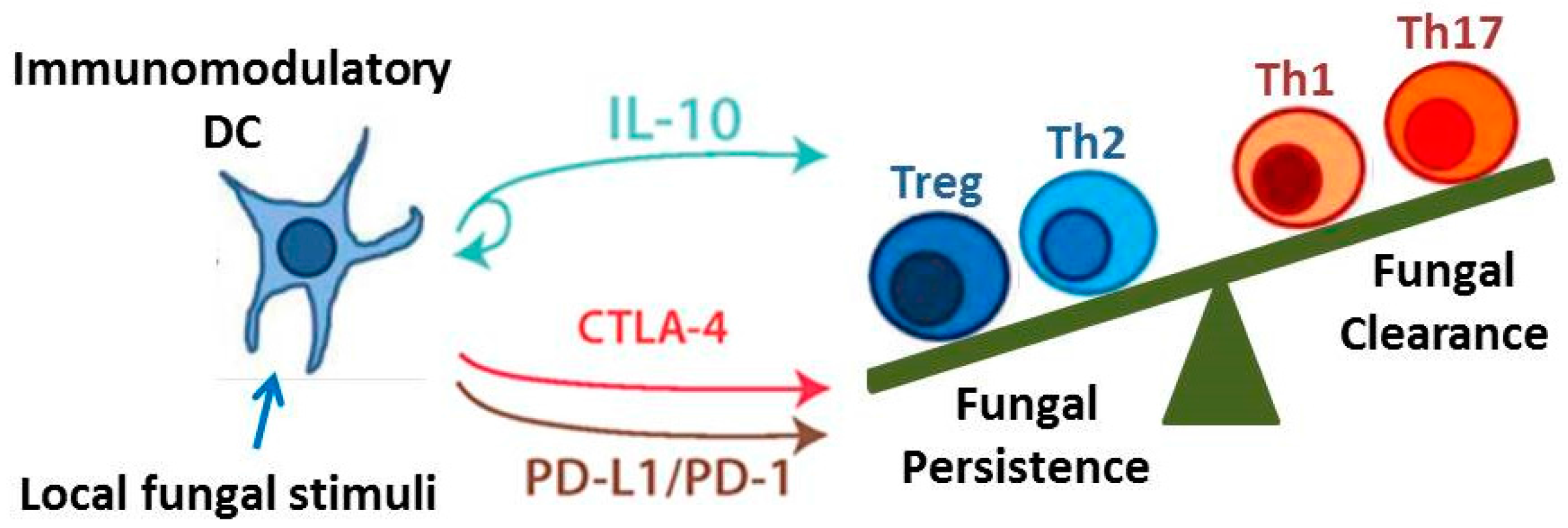{kind=link}
| Concept | Specific Topic | Fungi of Relevance | References |
|---|---|---|---|
| Cells | Dendritic Cells | A. fumigatus | [20,21,35] |
| C. albicans | [28,29,68] | ||
| C. neoformans | [9,30,31,32,33,36,37,38] | ||
| H. capsulatum | [34] | ||
| Regulatory T Cells | A. fumigatus | [64] | |
| C. albicans | [58,59,65,67,68,69,71] | ||
| C. neoformans | [61,62] | ||
| H. capsulatum | [63] | ||
| P. brasiliensis | [60,66,67] | ||
| Signaling Pathways | IL-10 | A. fumigatus | [84,87] |
| C. albicans | [83,85] | ||
| C. neoformans | [9,36,37,38,39,81] | ||
| H. capsulatum | [86] | ||
| PD-1 | C. albicans | [114,115] | |
| C. neoformans | [110] | ||
| H. capsulatum | [111] | ||
| P. brasiliensis | [112] | ||
| P. jirovecii | [93] | ||
| CTLA-4 | C. albicans | [115,131] | |
| C. neoformans | [129,130] | ||
| P. brasiliensis | [67,128] | ||
| Unique Circumstances | Protective Tolerance | A. fumigatus | [134,135,136] |
| C. albicans | [71,133] | ||
| Immune Restoration Inflammatory Syndrome | A. fumigatus | [156,157] | |
| C. immitis | [162,163] | ||
| C. neoformans | [143,144,145,146,147,148,149,150,151,152,153,155,159,160,161] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roussey, J.A.; Olszewski, M.A.; Osterholzer, J.J. Immunoregulation in Fungal Diseases. Microorganisms 2016, 4, 47. https://doi.org/10.3390/microorganisms4040047
Roussey JA, Olszewski MA, Osterholzer JJ. Immunoregulation in Fungal Diseases. Microorganisms. 2016; 4(4):47. https://doi.org/10.3390/microorganisms4040047
Chicago/Turabian StyleRoussey, Jonathan A., Michal A. Olszewski, and John J. Osterholzer. 2016. "Immunoregulation in Fungal Diseases" Microorganisms 4, no. 4: 47. https://doi.org/10.3390/microorganisms4040047
APA StyleRoussey, J. A., Olszewski, M. A., & Osterholzer, J. J. (2016). Immunoregulation in Fungal Diseases. Microorganisms, 4(4), 47. https://doi.org/10.3390/microorganisms4040047