
Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract


Abstract
:1. Introduction
2. Sources of H2S
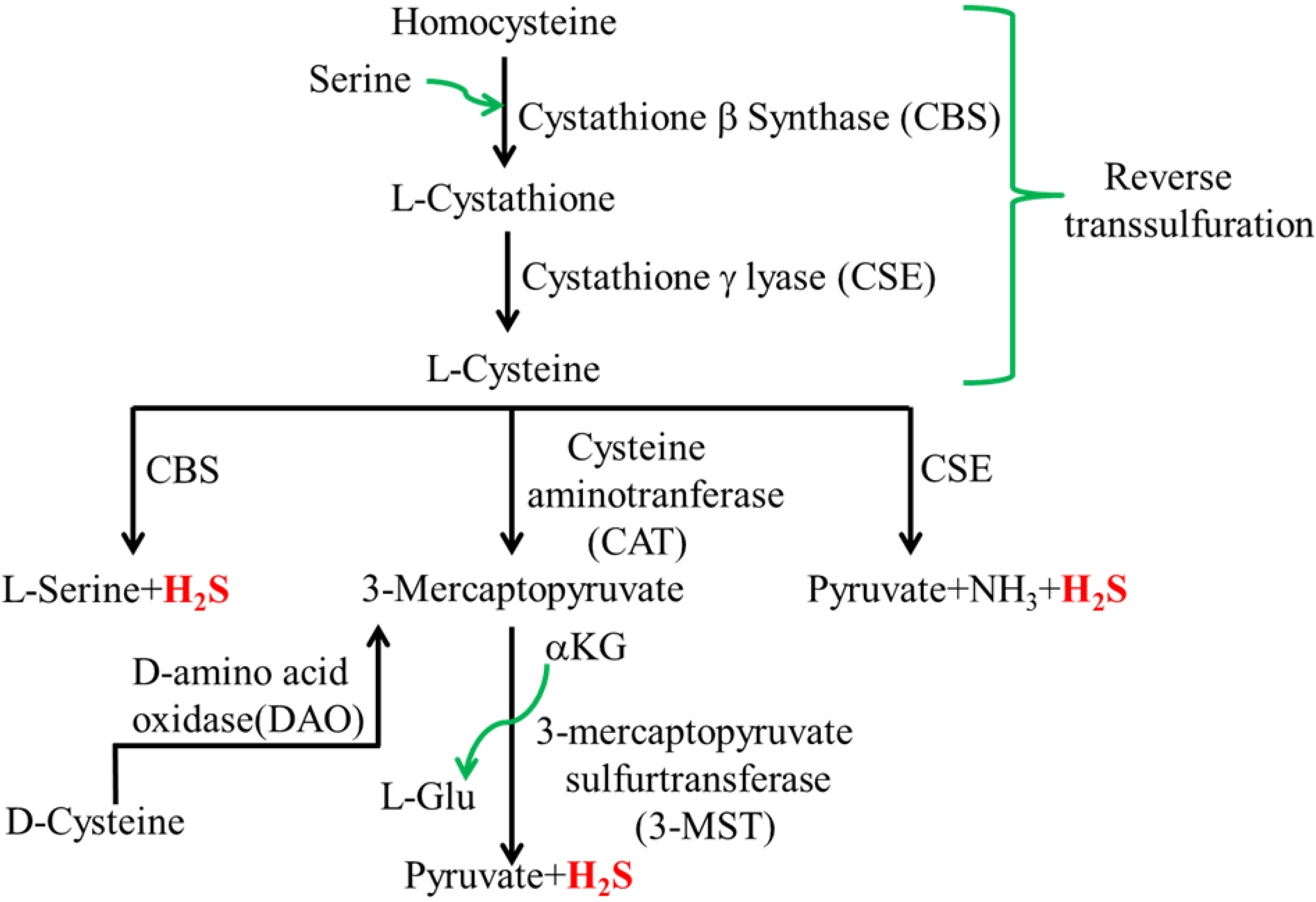
2.1. Mammalian H2S
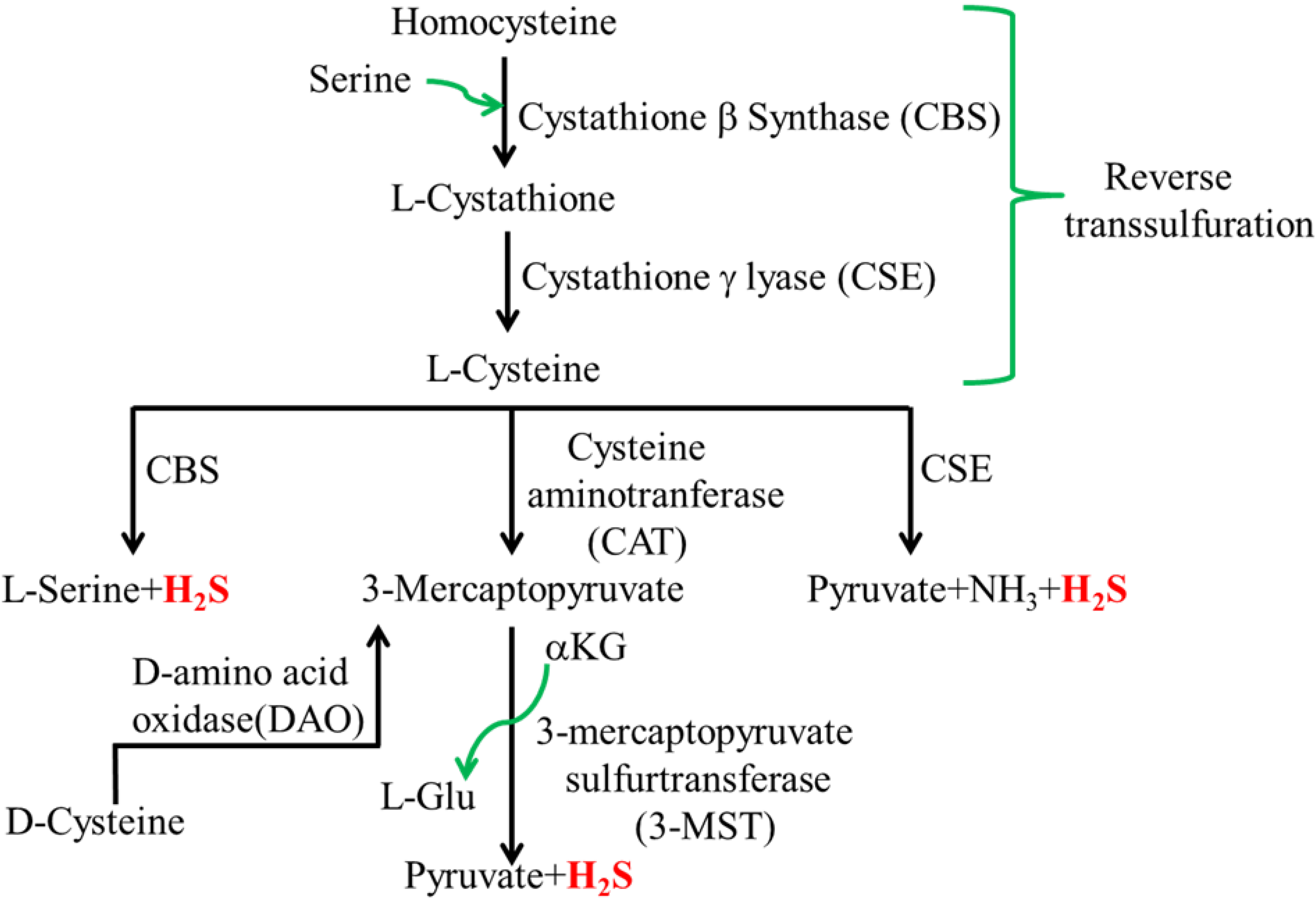
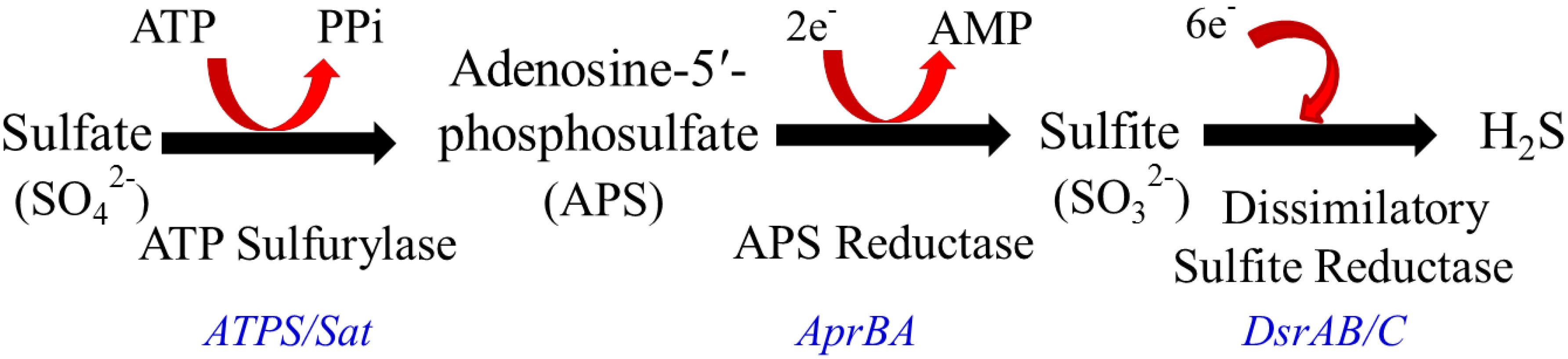
2.2. Intestinal H2S Production by Resident Microbes
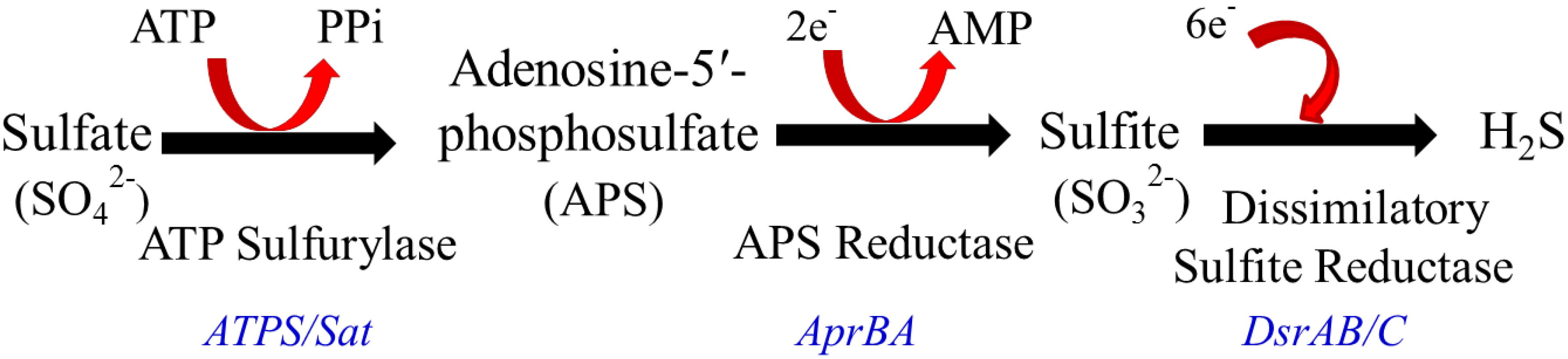
3. Impact of SRB, Bacterial Derived H2S and Lipopolysaccharide (LPS) on Host
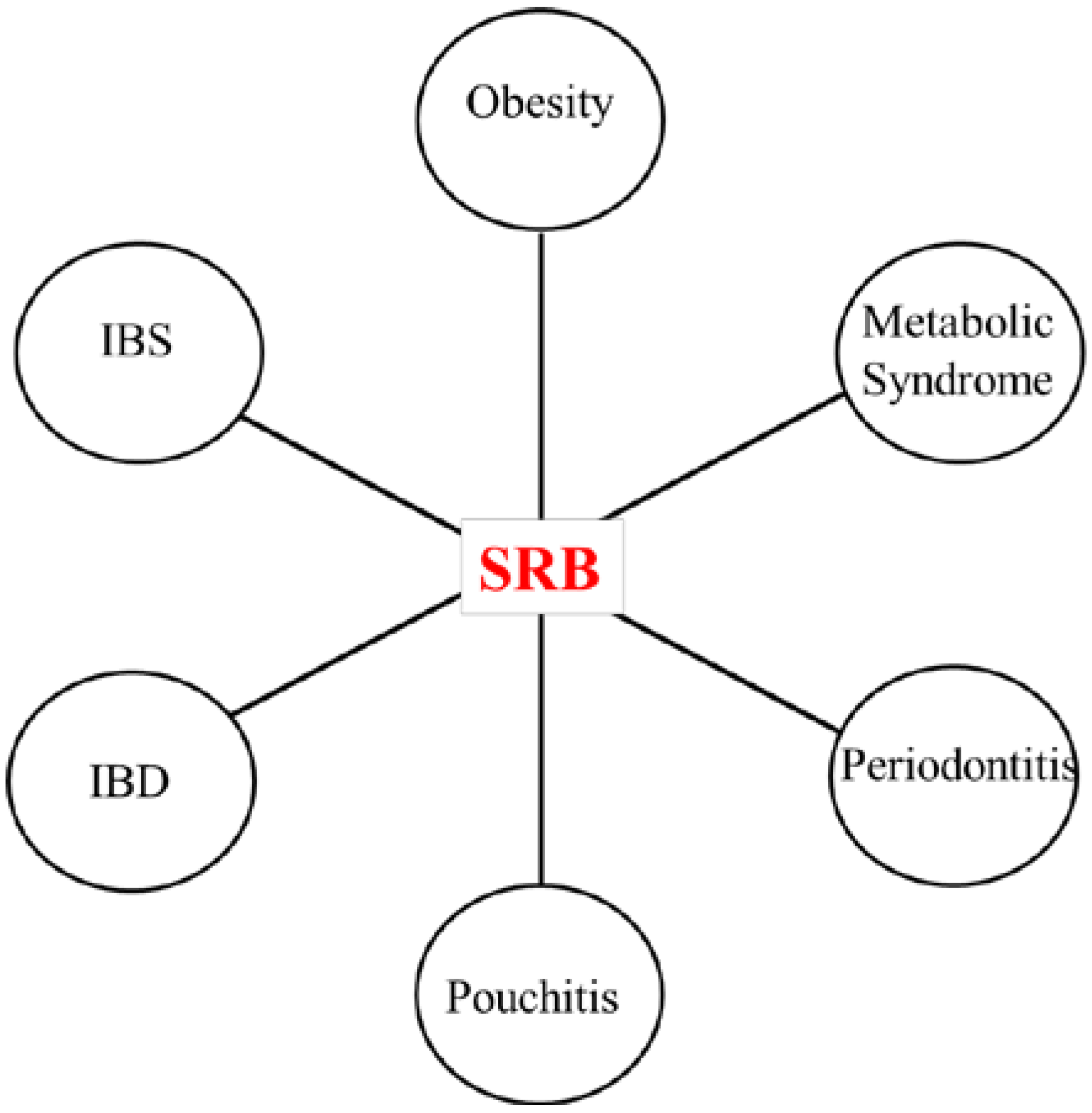
3.1. SRB and Inflammatory Diseases
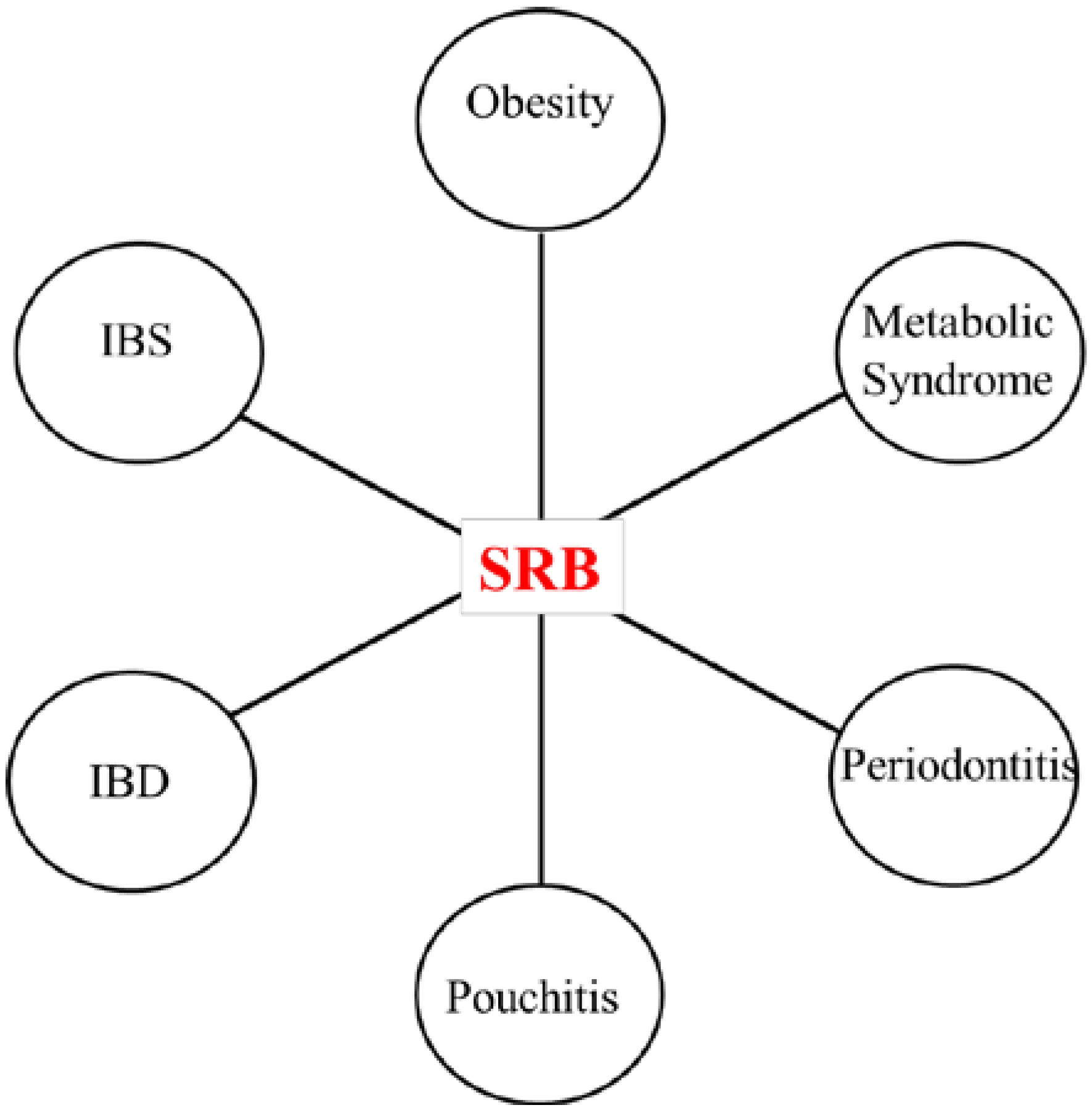
3.2. Effects of SRB Activity on Host
4. Effects of Host-Derived H2S on the Gastrointestinal System

{kind=link}
{kind=link}
{kind=link}
| Physiology | Disease |
|---|---|
| Protection against I/R Injury | Inflammatory Bowel Disease |
| Inflammation (Pro-/Anti-inflammatory) | Colitis |
| Motility (Excitation/Inhibitory) | Pouchitis |
| Nociception (Pro-/Anti-nociceptive) | Periodontitis |
| Colonic Secretion | Halitosis |
| Ulcer Healing | Sepsis |
| Antioxidant | Obesity |
| Apoptosis |
4.1. H2S in Ischemia/Reperfusion Injury
4.2. H2S in Intestinal Inflammation
4.3. H2S in Ulcer Healing
4.4. H2S in Intestinal Motility
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kfir, H.; Rimbrot, S.; Markel, A. Toxic effects of hydrogen sulfide: Experience with three simultaneous patients. QJM 2015. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Smith, D.R.; Canfield, D.V. A fatality caused by accidental production of hydrogen sulfide. Forensic. Sci. Int. 2001, 123, 211–214. [Google Scholar] [CrossRef]
- Snyder, J.W.; Safir, E.F.; Summerville, G.P.; Middleberg, R.A. Occupational fatality and persistent neurological sequelae after mass exposure to hydrogen sulfide. Am. J. Emerg. Med. 1995, 13, 199–203. [Google Scholar] [CrossRef]
- Yalamanchili, C.; Smith, M.D. Acute hydrogen sulfide toxicity due to sewer gas exposure. Am. J. Emerg. Med. 2008, 26, e5–e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed]
- Linden, D.R. Hydrogen sulfide signaling in the gastrointestinal tract. Antioxid. Redox Signal. 2014, 20, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.V.; Wallace, J.L. Hydrogen sulfide-based therapeutics and gastrointestinal diseases: Translating physiology to treatments. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G467–G473. [Google Scholar] [CrossRef] [PubMed]
- Medani, M.; Collins, D.; Docherty, N.G.; Baird, A.W.; O’Connell, P.R.; Winter, D.C. Emerging role of hydrogen sulfide in colonic physiology and pathophysiology. Inflamm. Bowel Dis. 2011, 17, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Mencarelli, A. Hydrogen sulfide in gastrointestinal and liver physiopathology. Inflamm. Allergy Drug Targets 2011, 10, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Magierowski, M.; Magierowska, K.; Kwiecien, S.; Brzozowski, T. Gaseous mediators nitric oxide and hydrogen sulfide in the mechanism of gastrointestinal integrity, protection and ulcer healing. Molecules 2015, 20, 9099–9123. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, G.; Szurszewski, J.H. Carbon monoxide, hydrogen sulfide, and nitric oxide as signaling molecules in the gastrointestinal tract. Gastroenterology 2014, 147, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Wang, R. Hydrogen sulfide-based therapeutics: Exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015, 14, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Stepién, P.P.; Pieniazek, N.J. The use of the l-serine sulfhydrylase assay for the estimation of cystathionine beta-synthase. Anal. Biochem. 1973, 54, 294–249. [Google Scholar] [CrossRef]
- Radcliffe, B.C.; Egan, A.R. A survey of methionine adenosyltransferase and cystathionine gamma-lyase activities in ruminant tissues. Aust. J. Biol. Sci. 1974, 27, 465–471. [Google Scholar] [PubMed]
- Peck, H.D. Enzymatic basis for assimilatory and dissimilatory sulfate reduction. J. Bacteriol. 1961, 82, 933–939. [Google Scholar] [PubMed]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Erickson, P.F.; Maxwell, I.H.; Su, L.J.; Baumann, M.; Glode, L.M. Sequence of cDNA for rat cystathionine gamma-lyase and comparison of deduced amino acid sequence with related Escherichia coli enzymes. Biochem. J. 1990, 269, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Searcy, D.G.; Lee, S.H. Sulfur reduction by human erythrocytes. J. Exp. Zool. 1998, 282, 310–322. [Google Scholar] [CrossRef]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from d-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, N.; Ito, T.; Kitamura, H.; Nishino, T. Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem. Cell Biol. 1998, 110, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R.; McKnight, G.W.; Dicay, M.S.; Coffin, C.S.; Ferraz, J.G.P.; Wallace, J.L. Hydrogen sulphide synthesis in the rat and mouse gastrointestinal tract. Dig. Liver Dis. 2010, 42, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Linden, D.R.; Sha, L.; Mazzone, A.; Stoltz, G.J.; Bernard, C.E.; Furne, J.K.; Levitt, M.D.; Farrugia, G.; Szurszewski, J.H. Production of the gaseous signal molecule hydrogen sulfide in mouse tissues. J. Neurochem. 2008, 106, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, P.; Yang, G.; Cao, Q.; Wang, R. The inhibitory role of hydrogen sulfide in airway hyperresponsiveness and inflammation in a mouse model of asthma. Am. J. Pathol. 2013, 182, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Schicho, R.; Krueger, D.; Zeller, F.; von Weyhern, C.W.H.; Frieling, T.; Kimura, H.; Ishii, I.; de Giorgio, R.; Campi, B.; Schemann, M. Hydrogen sulfide is a novel prosecretory neuromodulator in the Guinea-pig and human colon. Gastroenterology 2006, 131, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Mikami, Y.; Kimura, Y.; Nagahara, N.; Kimura, H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J. Biochem. 2009, 146, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; McCoy, K.D.; Wallace, J.L. Eukaryotic and prokaryotic contributions to colonic hydrogen sulfide synthesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G188–G193. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.E.; Hume, I.D. Contributions of microbes in vertebrate gastrointestinal tract to production and conservation of nutrients. Physiol Rev. 1998, 78, 393–427. [Google Scholar] [PubMed]
- Littman, D.R.; Pamer, E.G. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 2011, 10, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Nava, G.M.; Carbonero, F.; Croix, J.A.; Greenberg, E.; Gaskins, H.R. Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon. ISME J. 2012, 6, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Beerens, H.; Romond, C. Sulfate-reducing anaerobic bacteria in human feces. Am. J. Clin. Nutr. 1977, 30, 1770–1776. [Google Scholar] [PubMed]
- Gibson, G.R.; Macfarlane, G.T.; Cummings, J.H. Occurrence of sulphate-reducing bacteria in human faeces and the relationship of dissimilatory sulphate reduction to methanogenesis in the large gut. J. Appl. Bacteriol. 1988, 65, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Claesson, R.; Edlund, M.B.; Persson, S.; Carlsson, J. Production of volatile sulfur compounds by various Fusobacterium species. Oral Microbiol. Immunol. 1990, 5, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Yoshida, A.; Nagata, E.; Hoshino, T.; Oho, T.; Awano, S.; Takehara, T.; Ansai, T. Streptococcus anginosus l-cysteine desulfhydrase gene expression is associated with abscess formation in BALB/c mice. Mol. Oral Microbiol. 2011, 26, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kho, H.-S.; Chung, J.-W.; Chung, S.-C.; Kim, Y.-K. Volatile sulfur compounds produced by Helicobacter pylori. J. Clin. Gastroenterol. 2006, 40, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Metaxas, M.A.; Delwiche, E.A. The l-cysteine desulfhydrase of Escherichia coli. J. Bacteriol. 1955, 70, 735–737. [Google Scholar] [PubMed]
- Kredich, N.M.; Keenan, B.S.; Foote, L.J. The purification and subunit structure of cysteine desulfhydrase from Salmonella typhimurium. J. Biol. Chem. 1972, 247, 7157–7162. [Google Scholar] [PubMed]
- Carbonero, F.; Benefiel, A.C.; Alizadeh-Ghamsari, A.H.; Gaskins, H.R. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 2012, 3, 448. [Google Scholar] [CrossRef] [PubMed]
- Suarez, F.; Furne, J.; Springfield, J.; Levitt, M. Production and elimination of sulfur-containing gases in the rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 1998, 274, G727–G733. [Google Scholar]
- Furne, J.; Springfield, J.; Koenig, T.; DeMaster, E.; Levitt, M.D. Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: A specialized function of the colonic mucosa. Biochem. Pharmacol. 2001, 62, 255–259. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Pinkus, L.M.; Jakoby, W.B. Thiol S-methyltransferase: suggested role in detoxication of intestinal hydrogen sulfide. Biochem. Pharmacol. 1980, 29, 2885–2887. [Google Scholar] [CrossRef]
- Borchardt, R.T.; Cheng, C.F. Purification and characterization of rat liver microsomal thiol methyltransferase. Biochim. Biophys. Acta 1978, 522, 340–353. [Google Scholar] [CrossRef]
- Levitt, M.D.; Furne, J.; Springfield, J.; Suarez, F.; DeMaster, E. Detoxification of hydrogen sulfide and methanethiol in the cecal mucosa. J. Clin. Invest. 1999, 104, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; Amand, A.L.S.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.C. Small intestinal bacterial overgrowth: A framework for understanding irritable bowel syndrome. JAMA 2004, 292, 852–858. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Brummer, R.J. Alteration of the intestinal microbiota as a cause of and a potential therapeutic option in irritable bowel syndrome. Benef. Microbes 2014, 5, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.; Su, J.; Koprowski, S.; Hsu, A.; Tweddell, J.S.; Rafiee, P.; Gross, G.J.; Salzman, N.H.; Baker, J.E. Intestinal microbiota determine severity of myocardial infarction in rats. FASEB J. 2012, 26, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Borges Canha, M. Role of colonic microbiota in colorectal carcinogenesis: A systematic review. Rev. Esp. Enfermedades Dig. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Ohge, H.; Furne, J.K.; Springfield, J.; Sueda, T.; Madoff, R.D.; Levitt, M.D. The effect of antibiotics and bismuth on fecal hydrogen sulfide and sulfate-reducing bacteria in the rat. FEMS Microbiol. Lett. 2003, 228, 137–142. [Google Scholar] [CrossRef]
- Shatalin, K.; Shatalina, E.; Mironov, A.; Nudler, E. H2S: A universal defense against antibiotics in bacteria. Science 2011, 334, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Cummings, J.H.; Macfarlane, G.T. Growth and activities of sulphate-reducing bacteria in gut contents of healthy subjects and patients with ulcerative colitis. FEMS Microbiol. Ecol. 1991, 9, 103–111. [Google Scholar] [CrossRef]
- Loubinoux, J.; Bronowicki, J.-P.; Pereira, I.A.C.; Mougenel, J.-L.; Faou, A.E. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol. Ecol. 2002, 40, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.; Ellis, C.J.; Furne, J.K.; Springfield, J.; Levitt, M.D. Fecal hydrogen sulfide production in ulcerative colitis. Am. J. Gastroenterol. 1998, 93, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, M.C. The contribution of sulphate reducing bacteria and 5-aminosalicylic acid to faecal sulphide in patients with ulcerative colitis. Gut 2000, 46, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Jowett, S.L.; Seal, C.J.; Pearce, M.S.; Phillips, E.; Gregory, W.; Barton, J.R.; Welfare, M.R. Influence of dietary factors on the clinical course of ulcerative colitis: A prospective cohort study. Gut 2004, 53, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Edmond, L.M.; Hopkins, M.J.; Magee, E.A.; Cummings, J.H. The effect of 5-aminosalicylic acid-containing drugs on sulfide production by sulfate-reducing and amino acid-fermenting bacteria. Inflamm. Bowel Dis. 2003, 9, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E.; Duncan, A.; Kapaniris, O.; Millard, S. Reducing sulfur compounds of the colon impair colonocyte nutrition: implications for ulcerative colitis. Gastroenterology 1993, 104, 802–809. [Google Scholar] [PubMed]
- Den Hond, E.; Hiele, M.; Evenepoel, P.; Peeters, M.; Ghoos, Y.; Rutgeerts, P. In vivo butyrate metabolism and colonic permeability in extensive ulcerative colitis. Gastroenterology 1998, 115, 584–590. [Google Scholar] [CrossRef]
- Moore, J.; Babidge, W.; Millard, S.; Roediger, W. Colonic luminal hydrogen sulfide is not elevated in ulcerative colitis. Dig. Dis. Sci. 1998, 43, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Furne, J.K.; Suarez, F.L.; Ewing, S.L.; Springfield, J.; Levitt, M.D. Binding of hydrogen sulfide by bismuth does not prevent dextran sulfate-induced colitis in rats. Dig. Dis. Sci. 2000, 45, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Langendijk, P.S.; Hanssen, J.T.; van der Hoeven, J.S. Sulfate-reducing bacteria in association with human periodontitis. J. Clin. Periodontol. 2000, 27, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Persson, S. Hydrogen sulfide and methyl mercaptan in periodontal pockets. Oral Microbiol. Immunol. 1992, 7, 378–379. [Google Scholar] [CrossRef] [PubMed]
- Ohge, H.; Furne, J.K.; Springfield, J.; Rothenberger, D.A.; Madoff, R.D.; Levitt, M.D. Association between fecal hydrogen sulfide production and pouchitis. Dis. Colon Rectum 2005, 48, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Chassard, C.; Dapoigny, M.; Scott, K.P.; Crouzet, L.; Del’homme, C.; Marquet, P.; Martin, J.C.; Pickering, G.; Ardid, D.; Eschalier, A.; et al. Functional dysbiosis within the gut microbiota of patients with constipated-irritable bowel syndrome. Aliment. Pharmacol. Ther. 2012, 35, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, M.; Wang, S.; Han, R.; Cao, Y.; Hua, W.; Mao, Y.; Zhang, X.; Pang, X.; Wei, C.; et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 2010, 4, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, I.; Laleman, W.; Wauters, G.; Verhaegen, J. Desulfovibrio desulfuricans bacteremia in an immunocompromised host with a liver graft and ulcerative colitis. J. Clin. Microbiol. 2012, 50, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.; Citron, D.M.; Peraino, V.A.; Cross, S.A. Desulfovibrio desulfuricans bacteremia and review of human Desulfovibrio infections. J. Clin. Microbiol. 2003, 41, 2752–2754. [Google Scholar] [CrossRef] [PubMed]
- Koyano, S.; Tatsuno, K.; Okazaki, M.; Ohkusu, K.; Sasaki, T.; Saito, R.; Okugawa, S.; Moriya, K. A Case of Liver Abscess with Desulfovibrio Desulfuricans Bacteremia. Case Rep. Infect. Dis. 2015, 2015, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Guo, Z.; Kwok, L.; Ma, C.; Zhang, W.; Lv, Q.; Huang, W.; Zhang, H. Effect of oral consumption of probiotic Lactobacillus planatarum P-8 on fecal microbiota, SIgA, SCFAs, and TBAs of adults of different ages. Nutrition 2014, 30, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Sawin, E.A.; de Wolfe, T.J.; Aktas, B.; Stroup, B.M.; Murali, S.G.; Steele, J.L.; Ney, D.M. Glycomacropeptide is a prebiotic that reduces Desulfovibrio bacteria, increases cecal short chain fatty acids and is anti-inflammatory in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G590–G601. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Fei, N.; Pang, X.; Shen, J.; Wang, L.; Zhang, B.; Zhang, M.; Zhang, X.; Zhang, C.; Li, M.; et al. A gut microbiota-targeted dietary intervention for amelioration of chronic inflammation underlying metabolic syndrome. FEMS Microbiol. Ecol. 2014, 87, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.L.; Onnerfält, J.; Xu, J.; Molin, G.; Ahrné, S.; Thorngren-Jerneck, K. The microbiota of the gut in preschool children with normal and excessive body weight. Obesity 2012, 20, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; Devinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm. Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Dharmani, P.; Strauss, J.; Ambrose, C.; Allen-Vercoe, E.; Chadee, K. Fusobacterium nucleatum infection of colonic cells stimulates MUC2 mucin and tumor necrosis factor alpha. Infect. Immun. 2011, 79, 2597–2607. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Attene-Ramos, M.S.; Wagner, E.D.; Plewa, M.J.; Gaskins, H.R. Evidence that hydrogen sulfide is a genotoxic agent. Mol. Cancer Res. 2006, 4, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.E.; Thiennimitr, P.; Winter, M.G.; Butler, B.P.; Huseby, D.L.; Crawford, R.W.; Russell, J.M.; Bevins, C.L.; Adams, L.G.; Tsolis, R.M.; et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 2010, 467, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.L.; Clark, M.A. Tetrathionate reduction and production of hydrogen sulfide from thiosulfate. Microbiol. Rev. 1987, 51, 192–205. [Google Scholar] [PubMed]
- Clark, M.A.; Barrett, E.L. The phs gene and hydrogen sulfide production by Salmonella typhimurium. J. Bacteriol. 1987, 169, 2391–2397. [Google Scholar] [PubMed]
- Dzierzewicz, Z.; Szczerba, J.; Lodowska, J.; Wolny, D.; Gruchlik, A.; Orchel, A.; Weglarz, L. The role of Desulfovibrio desulfuricans lipopolysaccharides in modulation of periodontal inflammation through stimulation of human gingival fibroblasts. Arch. Oral Biol. 2010, 55, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Weglarz, L.; Dzierzewicz, Z.; Skop, B.; Orchel, A.; Parfiniewicz, B.; Wiśniowska, B.; Swiatkowska, L.; Wilczok, T. Desulfovibrio desulfuricans lipopolysaccharides induce endothelial cell IL-6 and IL-8 secretion and E-selectin and VCAM-1 expression. Cell. Mol. Biol. Lett. 2003, 8, 991–1003. [Google Scholar] [PubMed]
- Zhang-Sun, W.; Augusto, L.A.; Zhao, L.; Caroff, M. Desulfovibrio desulfuricans isolates from the gut of a single individual: Structural and biological lipid a characterization. FEBS Lett. 2015, 589, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Weglarz, L.; Dzierzewicz, Z.; Orchel, A.; Szczerba, J.; Jaworska-Kik, M.; Wilczok, T. Biological activity of Desulfovibrio desulfuricans lipopolysaccharides evaluated via interleukin-8 secretion by Caco-2 cells. Scand. J. Gastroenterol. 2003, 38, 73–79. [Google Scholar] [PubMed]
- Li, L.; Bhatia, M.; Zhu, Y.Z.; Zhu, Y.C.; Ramnath, R.D.; Wang, Z.J.; Anuar, F.B.M.; Whiteman, M.; Salto-Tellez, M.; Moore, P.K. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005, 19, 1196–1198. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Gonzalez, M.D.; Cheng, J.; Wu, M.; Ahern, P.P.; Gordon, J.I. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc. Natl. Acad. Sci. USA 2013, 110, 13582–13587. [Google Scholar] [CrossRef] [PubMed]
- Rubin, S.C.; Alava, P.; Zekker, I.; Laing, G.D.; van de Wiele, T. Arsenic thiolation and the role of sulfate-reducing bacteria from the human intestinal tract. Environ. Health Perspect. 2014, 122, 817–822. [Google Scholar]
- Wang, B.; Xu, H.; Wei, H.; Zeng, Z.; Xu, F. Oral administration of Bifidobacterim bifidum for modulating microflora, acid and bile resistance, and physiological indices in mice. Can. J. Microbiol. 2015, 61, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, J.; Hou, J.; Dong, Y.; Lu, Y.; Jin, L.; Cao, R.; Li, T.; Wu, J. Oral administration of recombinant Lactococcus lactis expressing HSP65 and tandemly repeated P277 reduces the incidence of type I diabetes in non-obese diabetic mice. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Arimatsu, K.; Kato, T.; Matsuda, Y.; Minagawa, T.; Takahashi, N.; Ohno, H.; Yamazaki, K. Oral Administration of P. gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Lyte, M.; Li, W.; Opitz, N.; Gaykema, R.P.; Goehler, L.E. Induction of anxiety-like behavior in mice during the initial stages of infection with the agent of murine colonic hyperplasia Citrobacter rodentium. Physiol. Behav. 2006, 89, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Wang, J.; Xiao, Y.; Bai, W.; Xie, L.; Shan, L.; Moore, P.K.; Ji, Y. GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. J. Biomed. Res. 2015, 29, 203–213. [Google Scholar] [PubMed]
- Sun, Y.-G.; Wang, X.-Y.; Chen, X.; Shen, C.-X.; Li, Y.-G. Hydrogen sulfide improves cardiomyocytes electrical remodeling post ischemia/reperfusion injury in rats. Int. J. Clin. Exp. Pathol. 2015, 8, 474–481. [Google Scholar] [PubMed]
- Sun, W.-H.; Liu, F.; Chen, Y.; Zhu, Y.-C. Hydrogen sulfide decreases the levels of ROS by inhibiting mitochondrial complex IV and increasing SOD activities in cardiomyocytes under ischemia/reperfusion. Biochem. Biophys. Res. Commun. 2012, 421, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Xu, Q.; Jia, J.; Ao, G.; Sun, Y.; Hu, L.; Alkayed, N.J.; Wang, C.; Cheng, J. Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem. Biophys. Res. Commun. 2015, 458, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xiao, J.; Kang, B.; Zhu, X.; Xin, N.; Wang, Z. PI3K/SGK1/GSK3β signaling pathway Is involved in inhibition of autophagy in neonatal rat cardiomyocytes exposed to hypoxia/reoxygenation by hydrogen sulfide. Exp. Cell Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Fukai, M.; Wakayama, K.; Ishikawa, T.; Kobayashi, N.; Kimura, T.; Yamashita, K.; Kamiyama, T.; Shimamura, T.; Taketomi, A.; et al. Hydrogen sulfide augments survival signals in warm ischemia and reperfusion of the mouse liver. Surg. Today 2015, 45, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Wang, F.; Chen, K.; Shen, M.; Dai, W.; Xu, L.; Zhang, Y.; Wang, C.; Li, J.; Yang, J.; et al. Hydrogen sulfide ameliorates ischemia/reperfusion-induced hepatitis by inhibiting apoptosis and autophagy pathways. Mediators Inflamm. 2014, 2014, 935251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fu, H.; Zhang, H.; Xu, F.; Zou, Z.; Liu, M.; Wang, Q.; Miao, M.; Shi, X. Hydrogen sulfide preconditioning protects rat liver against ischemia/reperfusion injury by activating Akt-GSK-3β signaling and inhibiting mitochondrial permeability transition. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Bos, E.M.; Wang, R.; Snijder, P.M.; Boersema, M.; Damman, J.; Fu, M.; Moser, J.; Hillebrands, J.-L.; Ploeg, R.J.; Yang, G.; et al. Cystathionine γ-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J. Am. Soc. Nephrol. 2013, 24, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Kim, J.I.; Park, J.-W.; Park, K.M. Hydrogen sulfide accelerates the recovery of kidney tubules after renal ischemia/reperfusion injury. Nephrol. Dial. Transplant 2015, 30, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Azizi, F.; Seifi, B.; Kadkhodaee, M.; Ahghari, P. Administration of hydrogen sulfide protects ischemia reperfusion-induced acute kidney injury by reducing the oxidative stress. Ir. J. Med. Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.-B.; Ji, X.; Zhu, S.-H.; Hu, Y.-M.; Zhang, L.-D.; Miao, X.-L.; Ma, R.-M.; Duan, M.-L.; Li, W.-Y. Hydrogen sulphide and mild hypothermia activate the CREB signaling pathway and prevent ischemia-reperfusion injury. BMC Anesthesiol. 2015, 15, 119. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Lu, Z.; Tao, L.; Yang, L.; Guo, Y.; Yang, Y.; Sun, X.; Ding, Q. ROS-Dependent Neuroprotective Effects of NaHS in Ischemia Brain Injury Involves the PARP/AIF Pathway. Cell. Physiol. Biochem. 2015, 36, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Pope, M.R.; Bukovnik, U.; Tomich, J.M.; Fleming, S.D. Small β2-glycoprotein I peptides protect from intestinal ischemia reperfusion injury. J. Immunol. 2012, 189, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bai, X.-B.; Shi, S.; Cao, Y.-X. Hydrogen sulfide protects from intestinal ischaemia-reperfusion injury in rats. J. Pharm. Pharmacol. 2009, 61, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.A.; Bulkley, G.B.; Granger, D.N.; Hamilton, S.R.; McCord, J.M. Ischemic injury in the cat small intestine: Role of superoxide radicals. Gastroenterology 1982, 82, 9–15. [Google Scholar] [PubMed]
- Schoenberg, M.H.; Fredholm, B.B.; Haglund, U.; Jung, H.; Sellin, D.; Younes, M.; Schildberg, F.W. Studies on the oxygen radical mechanism involved in the small intestinal reperfusion damage. Acta Physiol. Scand. 1985, 124, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Mohr, A.; Schoenberg, M.H.; Schildberg, F.W. Inhibition of lipid peroxidation by superoxide dismutase following regional intestinal ischemia and reperfusion. Res. Exp. Med. 1987, 187, 9–17. [Google Scholar] [CrossRef]
- Pan, H.; Chen, D.; Liu, B.; Xie, X.; Zhang, J.; Yang, G. Effects of sodium hydrosulfide on intestinal mucosal injury in a rat model of cardiac arrest and cardiopulmonary resuscitation. Life Sci. 2013, 93, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Henderson, P.W.; Weinstein, A.L.; Sung, J.; Singh, S.P.; Nagineni, V.; Spector, J.A. Hydrogen sulfide attenuates ischemia-reperfusion injury in in vitro and in vivo models of intestine free tissue transfer. Plast. Reconstr. Surg. 2010, 125, 1670–1978. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kalogeris, T.; Wang, M.; Zuidema, M.Y.; Wang, Q.; Dai, H.; Davis, M.J.; Hill, M.A.; Korthuis, R.J. Hydrogen sulfide preconditioning or neutrophil depletion attenuates ischemia-reperfusion-induced mitochondrial dysfunction in rat small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G44–G54. [Google Scholar] [CrossRef] [PubMed]
- Yusof, M.; Kamada, K.; Kalogeris, T.; Gaskin, F.S.; Korthuis, R.J. Hydrogen sulfide triggers late-phase preconditioning in postischemic small intestine by an NO- and p38 MAPK-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H868–H876. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, J.; Li, H.; Xue, M.; Ji, A.; Li, Y. Role of hydrogen sulfide in ischemia-reperfusion injury. Oxid. Med. Cell. Longev. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ferlito, M.; Wang, Q.; Fulton, W.B.; Colombani, P.M.; Marchionni, L.; Fox-Talbot, K.; Paolocci, N.; Steenbergen, C. Hydrogen sulfide [corrected] increases survival during sepsis: protective effect of CHOP inhibition. J. Immunol. 2014, 192, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhi, L.; Moore, P.K.; Bhatia, M. Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1193–L1201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhi, L.; Moochhala, S.; Moore, P.K.; Bhatia, M. Hydrogen sulfide acts as an inflammatory mediator in cecal ligation and puncture-induced sepsis in mice by upregulating the production of cytokines and chemokines via NF-kB. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L960–L971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Moochhala, S.M.; Bhatia, M. Endogenous hydrogen sulfide regulates inflammatory response by activating the erk pathway in polymicrobial sepsis. J. Immunol. 2008, 181, 4320–4331. [Google Scholar] [CrossRef] [PubMed]
- Zhi, L.; Ang, A.D.; Zhang, H.; Moore, P.K.; Bhatia, M. Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK-NF-kappa B pathway. J. Leukoc. Biol. 2007, 81, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Salto-Tellez, M.; Tan, C.-H.; Whiteman, M.; Moore, P.K. GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radic. Biol. Med. 2009, 47, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Peng, H.; Du, Q.; Lin, W.; Liu, Y. GYY4137, a hydrogen sulfide‑releasing molecule, inhibits the inflammatory response by suppressing the activation of nuclear factor‑kappa B and mitogen‑activated protein kinases in Coxsackie virus B3‑infected rat cardiomyocytes. Mol. Med. Rep. 2014, 11, 1837–1844. [Google Scholar] [PubMed]
- Liu, Z.; Han, Y.; Li, L.; Lu, H.; Meng, G.; Li, X.; Shirhan, M.; Peh, M.T.; Xie, L.; Zhou, S.; et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E(-/-) mice. Br. J. Pharmacol. 2013, 169, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fox, B.; Keeble, J.; Salto-Tellez, M.; Winyard, P.G.; Wood, M.E.; Moore, P.K.; Whiteman, M. The complex effects of the slow-releasing hydrogen sulfide donor GYY4137 in a model of acute joint inflammation and in human cartilage cells. J. Cell. Mol. Med. 2013, 17, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Li, L.; Rose, P.; Tan, C.-H.; Parkinson, D.B.; Moore, P.K. The Effect of Hydrogen Sulfide Donors on Lipopolysaccharide-Induced Formation of Inflammatory Mediators in Macrophages. Antioxid. Redox Signal. 2010, 12, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-W.; Zhu, J.; Zuo, S.; Zhang, J.-L.; Chen, Z.-Y.; Chen, G.-W.; Wang, X.; Pan, Y.-S.; Liu, Y.-C.; Wang, P.-Y. Protective effect of hydrogen sulfide on TNF-α and IFN-γ-induced injury of intestinal epithelial barrier function in Caco-2 monolayers. Inflamm. Res. 2015, 64, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liang, F.; Masood, W.S.; Yan, X. Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 S-sulfhydration, MAPK dependent anti-apoptosis and NF-κB dependent anti-inflammation pathway. Eur. J. Pharmacol. 2014, 725, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; Agbor, T.A.; Blackler, R.W.; Kim, J.J.; Khan, W.I.; Verdu, E.F.; Ferraz, J.G.; Wallace, J.L. Impaired hydrogen sulfide synthesis and IL-10 signaling underlie hyperhomocysteinemia-associated exacerbation of colitis. Proc. Natl. Acad. Sci. USA 2014, 111, 13559–13564. [Google Scholar] [CrossRef] [PubMed]
- Hirata, I.; Naito, Y.; Takagi, T.; Mizushima, K.; Suzuki, T.; Omatsu, T.; Handa, O.; Ichikawa, H.; Ueda, H.; Yoshikawa, T. Endogenous hydrogen sulfide is an anti-inflammatory molecule in dextran sodium sulfate-induced colitis in mice. Dig. Dis. Sci. 2011, 56, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Vong, L.; McKnight, W.; Dicay, M.; Martin, G.R. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 2009, 137, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Orlandi, S.; Mencarelli, A.; Caliendo, G.; Santagada, V.; Distrutti, E.; Santucci, L.; Cirino, G.; Wallace, J.L. Enhanced activity of a hydrogen sulphide-releasing derivative of mesalamine (ATB-429) in a mouse model of colitis. Br. J. Pharmacol. 2007, 150, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; Agbor, T.A.; Motta, J.-P.; Ferraz, J.G.; Wang, R.; Buret, A.G.; Wallace, J.L. Proresolution effects of hydrogen sulfide during colitis are mediated through hypoxia-inducible factor-1α. FASEB J. 2015, 29, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Motta, J.-P.; Flannigan, K.L.; Agbor, T.A.; Beatty, J.K.; Blackler, R.W.; Workentine, M.L.; da Silva, G.J.; Wang, R.; Buret, A.G.; Wallace, J.L. Hydrogen sulfide protects from colitis and restores intestinal microbiota biofilm and mucus production. Inflamm. Bowel Dis. 2015, 21, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Mariggiò, M.A.; Minunno, V.; Riccardi, S.; Santacroce, R.; de Rinaldis, P.; Fumarulo, R. Sulfide enhancement of PMN apoptosis. Immunopharmacol. Immunotoxicol. 1998, 20, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Dufton, N.; Natividad, J.; Verdu, E.F.; Wallace, J.L. Hydrogen sulfide and resolution of acute inflammation: A comparative study utilizing a novel fluorescent probe. Sci. Rep. 2012, 2, 499. [Google Scholar] [CrossRef] [PubMed]
- Blackler, R.W.; Motta, J.-P.; Manko, A.; Workentine, M.; Bercik, P.; Surette, M.G.; Wallace, J.L. Hydrogen sulphide protects against NSAID-enteropathy through modulation of bile and the microbiota. Br. J. Pharmacol. 2015, 172, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Devchand, P.R. Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal defense. Br. J. Pharmacol. 2005, 145, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Brancaleone, V.; Mitidieri, E.; Flower, R.J.; Cirino, G.; Perretti, M. Annexin A1 mediates hydrogen sulfide properties in the control of inflammation. J. Pharmacol. Exp. Ther. 2014, 351, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Dicay, M.; McKnight, W.; Martin, G.R. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007, 21, 4070–4076. [Google Scholar] [CrossRef] [PubMed]
- Mard, S.A.; Ashabi, A.; Badavi, M.; Dianat, M. Protective effects of vitamin B6 alone and in combination with l-cysteine and NaHS on ethanol and indomethacin-induced gastric lesions in mice. Iran. J. Basic Med. Sci. 2015, 18, 253–258. [Google Scholar] [PubMed]
- Medeiros, J.V.; Bezerra, V.H.; Gomes, A.S.; Barbosa, A.L.; Lima-Júnior, R.C.; Soares, P.M.; Brito, G.A.; Ribeiro, R.A.; Cunha, F.Q.; Souza, M.H. Hydrogen sulfide prevents ethanol-induced gastric damage in mice: role of ATP-sensitive potassium channels and capsaicin-sensitive primary afferent neurons. J. Pharmacol. Exp. Ther. 2009, 330, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Piña, A.E.; Tapia-Alvarez, G.R.; Navarrete, A. Inhibition of endogenous hydrogen sulfide synthesis by PAG protects against ethanol-induced gastric damage in the rat. Eur. J. Pharmacol. 2010, 630, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Mard, S.A.; Neisi, N.; Solgi, G.; Hassanpour, M.; Darbor, M.; Maleki, M. Gastroprotective effect of NaHS against mucosal lesions induced by ischemia-reperfusion injury in rat. Dig. Dis. Sci. 2012, 57, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Liu, L.; Zou, J.; Qiao, W.; Liu, H.; Qi, Y.; Yan, C. Protective effect of endogenous hydrogen sulfide against oxidative stress in gastric ischemia-reperfusion injury. Exp. Ther. Med. 2013, 5, 689–694. [Google Scholar] [PubMed]
- Flannigan, K.L.; Ferraz, J.G.; Wang, R.; Wallace, J.L. Enhanced synthesis and diminished degradation of hydrogen sulfide in experimental colitis: A site-specific, pro-resolution mechanism. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Lou, L.-X.; Geng, B.; Du, J.-B.; Tang, C.-S. Hydrogen sulphide-induced hypothermia attenuates stress-related ulceration in rats. Clin. Exp. Pharmacol. Physiol. 2008, 35, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Tang, G.; Cao, K.; Wu, L.; Wang, R. Molecular mechanism for H(2)S-induced activation of K(ATP) channels. Antioxid. Redox Signal. 2010, 12, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cui, J.; Song, C.-J.; Bian, J.-S.; Sparatore, A.; Del Soldato, P.; Wang, X.-Y.; Yan, C.-D. H(2)S-releasing aspirin protects against aspirin-induced gastric injury via reducing oxidative stress. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Antonelli, E.; Distrutti, E.; Rizzo, G.; Mencarelli, A.; Orlandi, S.; Zanardo, R.; Renga, B.; Di Sante, M.; Morelli, A.; et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology 2005, 129, 1210–1224. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Gil, V.; Gallego, D.; Jiménez, M. Effects of inhibitors of hydrogen sulphide synthesis on rat colonic motility. Br. J. Pharmacol. 2011, 164, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Duenes, J.A.; Sarr, M.G. Role of hydrogen sulfide as a gasotransmitter in modulating contractile activity of circular muscle of rat jejunum. J. Gastrointest. Surg. 2012, 16, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Kasparek, M.S.; Linden, D.R.; Farrugia, G.; Sarr, M.G. Hydrogen sulfide modulates contractile function in rat jejunum. J. Surg. Res. 2012, 175, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Gallego, D.; Clavé, P.; Donovan, J.; Rahmati, R.; Grundy, D.; Jiménez, M.; Beyak, M.J. The gaseous mediator, hydrogen sulphide, inhibits in vitro motor patterns in the human, rat and mouse colon and jejunum. Neurogastroenterol. Motil. 2008, 20, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Gil, V.; Parsons, S.; Gallego, D.; Huizinga, J.; Jimenez, M. Effects of hydrogen sulphide on motility patterns in the rat colon. Br. J. Pharmacol. 2013, 169, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Shafigullin, M.Y.; Zefirov, R.A.; Sabirullina, G.I.; Zefirov, A.L.; Sitdikova, G.F. Effects of a hydrogen sulfide donor on spontaneous contractile activity of rat stomach and jejunum. Bull. Exp. Biol. Med. 2014, 157, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Wu, L.; Wang, R. Interaction of hydrogen sulfide with ion channels. Clin. Exp. Pharmacol. Physiol. 2010, 37, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Teague, B.; Asiedu, S.; Moore, P.K. The smooth muscle relaxant effect of hydrogen sulphide in vitro: Evidence for a physiological role to control intestinal contractility. Br. J. Pharmacol. 2002, 137, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Quan, X.; Luo, H.; Liu, Y.; Xia, H.; Chen, W.; Tang, Q. Hydrogen sulfide regulates the colonic motility by inhibiting both l-type calcium channels and BKCa channels in smooth muscle cells of rat colon. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cutillas, M.; Gil, V.; Mañé, N.; Clavé, P.; Gallego, D.; Martin, M.T.; Jimenez, M. Potential role of the gaseous mediator hydrogen sulphide (H2S) in inhibition of human colonic contractility. Pharmacol. Res. 2015, 93, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Yamane, S.; Kanno, T.; Nakamura, H.; Fujino, H.; Murayama, T. Hydrogen sulfide-mediated regulation of contractility in the mouse ileum with electrical stimulation: roles of l-cysteine, cystathionine β-synthase, and K+ channels. Eur. J. Pharmacol. 2014, 740, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Yamane, S.; Nomura, R.; Yanagihara, M.; Nakamura, H.; Fujino, H.; Matsumoto, K.; Horie, S.; Murayama, T. l-cysteine/d,l-homocysteine-regulated ileum motility via system L and B°,+ transporter: Modification by inhibitors of hydrogen sulfide synthesis and dietary treatments. Eur. J. Pharmacol. 2015, 764, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Li, J.; Gong, L.; Xu, X.; Han, T.; Ye, Y.; Che, T.; Luo, Y.; Li, J.; Zhan, R.; et al. H2 S modulates duodenal motility in male rats via activating TRPV1 and K(ATP) channels. Br. J. Pharmacol. 2014, 171, 1534–1550. [Google Scholar] [CrossRef] [PubMed]
- Patacchini, R.; Santicioli, P.; Giuliani, S.; Maggi, C.A. Pharmacological investigation of hydrogen sulfide (H2S) contractile activity in rat detrusor muscle. Eur. J. Pharmacol. 2005, 509, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Trevisani, M.; Patacchini, R.; Nicoletti, P.; Gatti, R.; Gazzieri, D.; Lissi, N.; Zagli, G.; Creminon, C.; Geppetti, P.; Harrison, S. Hydrogen sulfide causes vanilloid receptor 1-mediated neurogenic inflammation in the airways. Br. J. Pharmacol. 2005, 145, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Kodela, R.; Nath, N.; Dastagirzada, Y.M.; Velázquez-Martínez, C.A.; Boring, D.; Kashfi, K. Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: A general property and evidence of a tissue type-independent effect. Biochem. Pharmacol. 2012, 83, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Caliendo, G.; Santagada, V.; Cirino, G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). Br. J. Pharmacol. 2010, 159, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.B.; Lin, H.C. Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms 2015, 3, 866-889. https://doi.org/10.3390/microorganisms3040866
Singh SB, Lin HC. Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms. 2015; 3(4):866-889. https://doi.org/10.3390/microorganisms3040866
Chicago/Turabian StyleSingh, Sudha B., and Henry C. Lin. 2015. "Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract" Microorganisms 3, no. 4: 866-889. https://doi.org/10.3390/microorganisms3040866
APA StyleSingh, S. B., & Lin, H. C. (2015). Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms, 3(4), 866-889. https://doi.org/10.3390/microorganisms3040866