
C1-Pathways in Methyloversatilis universalis FAM5: Genome Wide Gene Expression and Mutagenesis Studies


Abstract
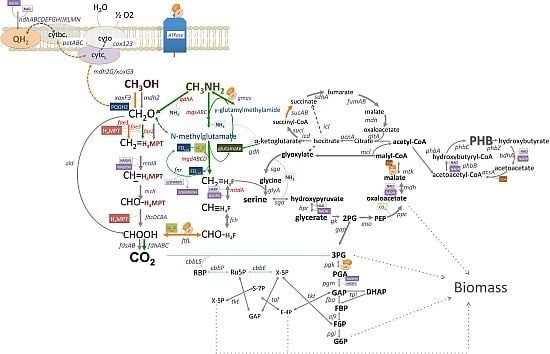
:
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids and Culture Conditions

{kind=link}
{kind=link}
| Strain | Description | Succinate | Methanol | Methylamine | Succinate + C1 * |
|---|---|---|---|---|---|
| FAM5 | Wild type, forms aggregates in liquid culture, KanSTetSCmR | ++++ | +++ | ++++ | ++++ |
| FAM5E | Trait of FAM5 which does not form aggregates in liquid culture, KanSTetSCmR | ++++ | +++ | ++++ | ++++ |
| ΔmgdD | FAM5-ΔmgdD:kan, KanRTetSCmR | ++++ | +++ | - | NT |
| ΔmgsC | FAM5-ΔmgsC:kan, KanRTetSCmR | ++++ | +++ | - | NT |
| Δfae1 | FAM5E-Δfae1:kan, KanRTetSCmR | ++++ | - | - | NT |
| Δfae2 | FAM5E-Δfae2:kan, KanRTetSCmR | ++++ | +++ | ++++ | NT |
| Δfae3 | FAM5E-Δfae3:kan, KanRTetSCmR | ++++ | +++ | ++++ | NT |
| ΔmtdA | FAM5E-ΔmtdA:kan, KanRTetSCmR | ++++ | - | - | +++ (1) |
| ΔqhpA | FAM5-ΔqhpA, KanSTetSCmR | ++++ | +++ | ++++ | NT |
| Δfae1ΔqhpA | FAM5-ΔqhpAΔfae1:kan, KanRTetSCmR | ++++ | - | ++ | NT |
| Δfae2ΔqhpA | FAM5-ΔqhpAΔfae2:kan, KanRTetSCmR | ++++ | +++ | ++++ | NT |
| Δfae3ΔqhpA | FAM5-ΔqhpAΔfae3:kan, KanRTetSCmR | ++++ | +++ | ++++ | NT |
| Δfae1RΔqhpAΔfae3 | FAM5E-ΔqhpA Δfae1Δfae3:kan, KanRTetSCmR | ++++ | - | - | NT |
| Δfae1R | Revertant of FAM5E-Δfae1, KanSTetSCmR | ++++ | - | ++ | ++++ (2) |
| Δfae1Δfae3 | FAM5-Δfae1Δfae3:kan, KanRTetSCmR | ++++ | - | - | NT |
| Δfae1RΔ fae3 | FAM5-Δfae1Δfae3, KanSTetSCmR | ++++ | - | ++ | ++++ (2) |
| Δfae1Δ fae2 | FAM5-Δfae1Δfae2:kan, KanRTetSCmR | ++++ | +++ | ++++ | NT |
| Δfae3 | FAM5E-Δfae3, KanSTetSCmR | ++++ | +++ | ++++ | NT |
| Δfae3Δfae2 | FAM5E-Δfae3 Δfae2:kan, KanRTetSCmR | ++++ | +++ | ++++ | NT |
| Δfae1Δfae2Δfae3 | FAM5E-Δfae1Δfae3Δfae2:kan, KanRTetSCmR | ++++ | - | - | NT |
| Strain/Plasmid | Markers/Description | Reference/Source |
|---|---|---|
| Escherichia coli One Shot TOP 10 | F-mcrA Δ (mrr-hsdRMS-mcrBC) Φ80 lacZΔM15 ΔlacX74 recA1 araD139 Δ (ara-leu)7697 galU galK rpsL StrR endA1 nupG | Invitrogen |
| Escherichia coli S17-1 | recA pro hsdR RP4-2-Tc:Mu-Km:Tn7 TmpR SpcRStrR | [28] |
| pCR2.1 | KanR, AmpR | Invitrogen |
| pDrive | KanR, AmpR | Qiagen |
| pCM184 | Broad-host-range allelic exchange vector, KanR, TetR | [27] |
| pCM157 | Cre/LoxP, TetR | [27] |
| pMgdD | pCM184 with mgdD upstream and downstream flanks | This study |
| pMgsC | pCM184 with mgsC upstream and downstream flanks | This study |
| pFAE1 | pCM184 with fae1 upstream and downstream flanks | This study |
| pFAE2 | pCM184 with fae2 upstream and downstream flanks | This study |
| pFAE3 | pCM184 with fae3 upstream and downstream flanks | This study |
| pQHNDH1 | pCM184 with qhpA upstream and downstream flanks | This study |
| pQHNDH2 | pCM184 with qhpX upstream and downstream flanks | This study |
| pMtdA | pCM184 with mtdA upstream and downstream flanks | This study |
2.2. DNA Manipulations
2.3. RNA-seq Experiments
2.4. Enzyme Assays
2.5. Accession Numbers
3. Results
3.1. Gene Expression Profiles: Methanol vs. Methylamine
| Gene ID (Old) | Gene ID (New) | Gene Product | Methanol (RPKM) * | Methylamine (RPKM) * | Fold Change | q-Value |
|---|---|---|---|---|---|---|
| Methylamine oxidation | ||||||
| METUNv1_760110 | METUNv2_580110 | Gmas, Gamma glutamylmethylamide synthetase | 2961.80 | 19,440.25 | 6.56 | 0.65 |
| METUNv1_760111 | METUNv2_580111 | MgsA, N-methyl glutamate synthase subunit A | 2727.43 | 22,221.30 | 8.15 | 0.73 |
| METUNv1_760112 | METUNv2_580112 | MgsB, N-methyl glutamate synthase subunit B | 2680.76 | 19,852.55 | 7.41 | 0.69 |
| METUNv1_760113 | METUNv2_580113 | MgsC, N-methyl glutamate synthase subunit C | 4221.56 | 53,956.55 | 12.78 | 0.67 |
| METUNv1_760114 | METUNv2_580114 | MgdA, N-methyl glutamate dehydrogenase/oxidoreductase subunit A | 1414.90 | 13,014.45 | 9.20 | 0.74 |
| METUNv1_760115 | METUNv2_580115 | MgdB, N-methyl glutamate dehydrogenase/oxidoreductase subunit B | 388.00 | 3906.31 | 10.07 | 0.74 |
| METUNv1_760116 | METUNv2_580116 | MgdC, N-methyl glutamate dehydrogenase/oxidoreductase subunit C | 3389.10 | 26,611.05 | 7.85 | 0.75 |
| METUNv1_760117 | METUNv2_580117 | MgdD, N-methyl glutamate dehydrogenase/oxidoreductase subunit D | 701.94 | 7962.74 | 11.34 | 0.71 |
| METUNv1_760127 | METUNv2_580127 | QhpB, Quinohemoprotein amine dehydrogenase, beta subunit | 420.06 | 4164.18 | 9.91 | 0.68 |
| METUNv1_760128 | METUNv2_580128 | QhpA, Quinohemoprotein amine dehydrogenase, alpha subunit | 733.76 | 7390.84 | 10.07 | 0.65 |
| METUNv1_760130 | METUNv2_580130 | QhpC, Quinohemoprotein amine dehydrogenase, SAM-radical dependent activating subunit | 380.86 | 4283.06 | 11.25 | 0.00 |
| Methanol oxidation | ||||||
| METUNv1_770214 | METUNv2_590217 | XoxF1, PQQ-linked dehydrogenase | 2023.28 | 3380.16 | 1.67 | 0.50 |
| METUNv1_770216 | METUNv2_590218 | XoxF2, PQQ-linked dehydrogenase | 10,962.17 | 16,741.70 | 1.53 | 0.05 |
| METUNv1_590046 | METUNv2_420045 | XoxF3, PQQ-dependent dehydrogenase | 1213.63 | 659.01 | −1.84 | 0.62 |
| METUNv1_590042 | METUNv2_420041 | XoxJ3, Extracellular solute-binding protein family 3 | 4399.57 | 1403.51 | −3.13 | 0.05 |
| METUNv1_590043 | METUNv2_420042 | XoxG3, Cytochrome c class I | 3156.11 | 1089.89 | −2.90 | 0.30 |
| METUNv1_590049 | METUNv2_420048 | Mdh2, PQQ-dependent methanol/ethanol dehydrogenase | 48,100.35 | 34,793.20 | −1.38 | 0.77 |
| METUNv1_590050 | METUNv2_420049 | Mdh2J, Extracellular solute-binding protein family | 2486.88 | 903.00 | −2.75 | 0.69 |
| METUNv1_590051 | METUNv2_420050 | Mdh2G, cytochrome c-type protein | 1732.91 | 595.39 | −2.91 | 0.58 |
| Formaldehyde oxidation | ||||||
| METUNv1_580096 | METUNv2_410093 | Fae 1, Formaldehyde-activating enzyme | 18,235.00 | 11,635.85 | −1.57 | 0.82 |
| METUNv1_660037 | METUNv2_480039 | Fae 2, Formaldehyde-activating enzyme | 561.52 | 642.50 | 1.14 | 0.89 |
| METUNv1_700516 | METUNv2_520523 | Fae 3, Formaldehyde activating enzyme | 584.51 | 6861.86 | 11.74 | 0.00 |
| METUNv1_590006 | METUNv2_420006 | Orf 9, Involved in biosynthesis of tetrahydromethanopterin. Essential for formaldehyde oxidation. | 430.06 | 187.51 | −2.29 | 0.21 |
| METUNv1_580095 | METUNv2_410092 | Orf 7, Involved in tetrahydromethanopterin-linked formaldehyde oxidation. | 371.25 | 549.26 | 1.48 | 0.75 |
| METUNv1_580094 | METUNv2_410091 | Orf 5, Involved in biosynthesis of tetrahydromethanopterin | 489.69 | 493.89 | 1.01 | 0.93 |
| METUNv1_580093 | METUNv2_410090 | Mch, Methenyltetrahydromethanopterin cyclohydrolase | 620.26 | 736.89 | 1.19 | 0.87 |
| METUNv1_580092 | METUNv2_410089 | OrfY, Involved in tetrahydromethanopterin C1 transfer. | 896.40 | 819.12 | −1.09 | 0.94 |
| METUNv1_580091 | METUNv2_410088 | MtdB, NAD-dependent methylenetetrahydromethanopterin dehydrogenase | 1295.29 | 1379.77 | 1.07 | 0.88 |
| METUNv1_580088 | METUNv2_410086 | FhcB, Formyltransferase/hydrolase complex subunit B | 587.63 | 515.00 | −1.14 | 0.92 |
| METUNv1_580087 | METUNv2_410085 | FhcA, Formyltransferase/hydrolase complex subunit A | 766.77 | 744.38 | −1.03 | 0.93 |
| METUNv1_580086 | METUNv2_410084 | FhcD, Formyltransferase/hydrolase complex subunit D | 598.76 | 547.64 | −1.09 | 0.94 |
| METUNv1_580085 | METUNv2_410083 | FhcC, Formyltransferase/hydrolase complex subunit C | 634.50 | 467.32 | −1.36 | 0.88 |
| METUNv1_590040 | METUNv2_420039 | Ald, Tungsten-containing aldehyde ferredoxin oxidoreductase | 408.13 | 155.57 | −2.62 | 0.17 |
| METUNv1_490013 | METUNv2_320014 | AldB, Aldehyde dehydrogenase B | 2762.93 | 2224.63 | −1.24 | 0.94 |
| Formate oxidation | ||||||
| METUNv1_770385 | METUNv2_590384 | Fdh, Putative formate dehydrogenase subunit A | 634.02 | 668.94 | 1.06 | 0.92 |
| METUNv1_700257 | METUNv2_520260 | FdhD, NAD-linked formate dehydrogenase delta subunit | 38.00 | 76.63 | 2.02 | 0.55 |
| METUNv1_700258 | METUNv2_520261 | FdhC, Formate dehydrogenase, accessory protein | 751.50 | 1024.07 | 1.36 | 0.74 |
| METUNv1_700259 | METUNv2_520262 | FdhA, NAD-dependent formate dehydrogenase alpha subunit | 3720.44 | 5479.72 | 1.47 | 0.39 |
| METUNv1_700260 | METUNv2_520263 | FdhB, NAD-dependent formate dehydrogenase beta subunit | 2758.00 | 4434.84 | 1.61 | 0.46 |
| METUNv1_700261 | METUNv2_520264 | FdhG, NAD-dependent formate dehydrogenase gamma subunit | 1127.75 | 1882.41 | 1.67 | 0.67 |
| METUNv1_700262 | METUNv2_520265 | FdhR, Formate dehydrogenase regulator | 210.25 | 231.00 | 1.10 | 0.90 |
| METUNv1_570005 | METUNv2_400021 | FdsA, NAD-dependent, tungsten-containing formate dehydrogenase alpha subunit | 1438.46 | 1527.51 | 1.06 | 0.91 |
| METUNv1_570006 | METUNv2_400022 | FdsB, NAD-dependent, tungsten-containing formate dehydrogenase beta subunit | 951.26 | 1018.18 | 1.07 | 0.90 |
| H4F-pathway/Serine cycle/Glyoxylate shunt | ||||||
| METUNv1_460318 | METUNv2_290319 | FtfL, formate-tetrahydrofolate ligase/synthetase | 4729.66 | 1674.32 | −2.82 | 0.54 |
| METUNv1_460309 | METUNv2_290310 | Fch, Methenyltetrahydrofolate cyclohydrolase | 2091.39 | 1541.64 | −1.36 | 0.90 |
| METUNv1_460310 | METUNv2_290311 | MdtA, NADP-dependent methylenetetrahydrofolate dehydrogenase | 4363.44 | 2973.78 | −1.47 | 0.93 |
| METUNv1_460311 | METUNv2_290312 | Hpr, Hydroxypyruvate reductase, NAD(P)H-dependent. | 3620.17 | 3220.89 | −1.12 | 0.89 |
| METUNv1_460312 | METUNv2_290313 | Sga, Serine-glyoxylate aminotransferase | 10,590.93 | 8570.41 | −1.24 | 0.79 |
| METUNv1_460313 | METUNv2_290314 | GlyA, Serine hydroxymethyltransferase | 6313.10 | 5292.54 | −1.19 | 0.87 |
| METUNv1_460314 | METUNv2_290315 | MtkA, Malate thiokinase large subunit | 4955.01 | 6292.07 | 1.27 | 0.73 |
| METUNv1_460315 | METUNv2_290316 | MtkB, Malate thiokinase small subunit | 5081.45 | 5363.84 | 1.06 | 0.73 |
| METUNv1_460316 | METUNv2_290317 | Ppc1, phosphoenolpyruvate carboxylase | 2661.63 | 2768.19 | 1.04 | 0.86 |
| METUNv1_460317 | METUNv2_290318 | Mcl, malyl-CoA lyase | 3260.85 | 4275.94 | 1.31 | 0.58 |
| METUNv1_770329 | METUNv2_590331 | Gk, Glycerate kinase | 1197.01 | 1116.38 | −1.07 | 0.93 |
| METUNv1_770169 | METUNv2_590170 | Ppc2, Phosphoenolpyruvate carboxylase | 1034.15 | 1074.00 | 1.04 | 0.91 |
| METUNv1_460302 | METUNv2_290303 | Eno, Enolase | 2095.13 | 1936.51 | −1.08 | 0.92 |
| METUNv1_710053 | METUNv2_530053 | Pgm, Phosphoglyceromutase | 470.57 | 517.14 | 1.10 | 0.90 |
| METUNv1_620020 | METUNv2_450021 | Ms, Malate synthase A | 353.94 | 269.76 | −1.31 | 0.89 |
| METUNv1_620018 | METUNv2_450018 | Icl, Isocitrate lyase | 12,174.92 | 8389.98 | −1.45 | 0.85 |
| CO2 Fixation (CBB cycle) | ||||||
| METUNv1_750044 | METUNv2_570044 | CbbR, RuBisCO operon transcriptional regulator | 195.62 | 189.12 | −1.03 | 0.93 |
| METUNv1_750045 | METUNv2_570045 | CbbL, Ribulose-1,5-bisphosphate carboxylase large subunit | 127.63 | 130.88 | 1.03 | 0.94 |
| METUNv1_750046 | METUNv2_570046 | CbbS, Ribulose bisphosphate carboxylase small subunit | 41.62 | 47.50 | 1.14 | 0.92 |
| METUNv1_750047 | METUNv2_570047 | CbxX, chromosomal AAA type ATPase | 8.75 | 11.94 | 1.36 | 0.89 |
| METUNv1_750048 | METUNv2_570048 | CbbY, haloacid dehalogenase | 7.25 | 14.56 | 2.01 | 0.73 |
| METUNv1_750049 | METUNv2_570049 | CbbE, Ribulose-phosphate 3-epimerase | 13.88 | 16.13 | 1.16 | 0.91 |
| METUNv1_750050 | METUNv2_570050 | Pgp, phosphoglycolate phosphatase | 12.50 | 13.00 | 1.04 | 0.94 |
| METUNv1_750051 | METUNv2_570051 | CbbF, Fructose-1,6-bisphosphatase | 26.75 | 30.51 | 1.14 | 0.92 |
| METUNv1_750052 | METUNv2_570052 | CbbP, Phosphoribulokinase | 26.69 | 32.00 | 1.20 | 0.92 |
| METUNv1_750053 | METUNv2_570053 | CbbT, Transketolase | 24.88 | 42.56 | 1.71 | 0.73 |
| METUNv1_750054 | METUNv2_570054 | CbbG, Glyceraldehyde-3-phosphate dehydrogenase | 17.50 | 19.56 | 1.12 | 0.93 |
| METUNv1_750055 | METUNv2_570055 | CbbA, Fructose-bisphosphate aldolase | 72.75 | 65.50 | −1.11 | 0.93 |
| METUNv1_700111 | METUNv2_520114 | CbbR, RuBisCO operon transcriptional regulator | 360.50 | 255.84 | −1.41 | 0.90 |
| METUNv1_760018 | METUNv2_580018 | CbbQ, Post-translational RubisCO activator | 16.25 | 19.38 | 1.19 | 0.91 |
| Sugar Phosphate Interconversions | ||||||
| METUNv1_470279 | METUNv2_300282 | Fbp, Fructose-1,6-bisphosphatase | 403.90 | 440.81 | 1.09 | 0.91 |
| METUNv1_700104 | METUNv2_520106 | Fba, Fructose-bisphosphate aldolase | 920.68 | 1094.83 | 1.19 | 0.85 |
| METUNv1_700105 | METUNv2_520107 | Pyk, Pyruvate kinase II | 807.00 | 786.28 | −1.03 | 0.92 |
| METUNv1_700106 | METUNv2_520108 | Pgk, Phosphoglycerate kinase | 693.63 | 684.62 | −1.01 | 0.92 |
| METUNv1_700107 | METUNv2_520109 | Gapdh, Glyceraldehyde 3-phosphate dehydrogenase | 1478.94 | 1536.51 | 1.04 | 0.90 |
| METUNv1_700108 | METUNv2_520110 | Tk, Transketolase | 982.15 | 730.45 | −1.34 | 0.89 |
| METUNv1_700109 | METUNv2_520111 | Prk, Phosphoribulokinase | 380.94 | 483.87 | 1.27 | 0.82 |
| METUNv1_700110 | METUNv2_520112 | Fbp3, Fructose-1,6-bisphosphatase | 902.64 | 846.62 | −1.07 | 0.94 |
| METUNv1_470090 | METUNv2_300091 | Pps, Phosphoenolpyruvate synthase | 1610.66 | 1360.78 | −1.18 | 0.94 |
| METUNv1_460093 | METUNv2_290089 | Pck, Phosphoenolpyruvate carboxykinase (GTP) | 242.99 | 191.39 | −1.27 | 0.89 |
| METUNv1_580038 | METUNv2_410037 | Pfk, Pyrophosphate-dependent phosphofructokinase | 866.70 | 1075.27 | 1.24 | 0.85 |
| METUNv1_580036 | METUNv2_410036 | Pyrophosphate-energized inorganic pyrophosphatase | 747.52 | 887.26 | 1.19 | 0.88 |
| METUNv1_580049 | METUNv2_410048 | Pyrophosphate phosphohydrolase | 555.12 | 602.37 | 1.09 | 0.89 |
| TCA cycle | ||||||
| METUNv1_700127 | METUNv2_520130 | E1 component of pyruvate dehydrogenase | 959.88 | 1210.51 | 1.26 | 0.80 |
| METUNv1_700126 | METUNv2_520129 | E2 component of pyruvate dehydrogenase | 217.00 | 302.25 | 1.39 | 0.77 |
| METUNv1_700125 | METUNv2_520128 | E3 component of pyruvate dehydrogenase | 482.26 | 555.08 | 1.15 | 0.88 |
| METUNv1_700186 | METUNv2_520190 | Succinyl-CoA synthetase beta subunit | 705.14 | 556.31 | −1.27 | 0.91 |
| METUNv1_700187 | METUNv2_520191 | Succinyl-CoA synthetase alpha subunit | 622.44 | 414.43 | −1.50 | 0.86 |
| METUNv1_470127 | METUNv2_300128 | Fumarate hydratase class I | 659.82 | 590.00 | −1.12 | 0.93 |
| METUNv1_460167 | METUNv2_290164 | Fumarate hydratase class II (fumarase C) | 185.50 | 141.26 | −1.31 | 0.91 |
| METUNv1_460003 | METUNv2_290002 | ME1, Malic Enzyme | 952.12 | 789.15 | −1.21 | 0.93 |
| METUNv1_520009 | METUNv2_350011 | Mdh, Malate dehydrogenase | 2673.56 | 1934.79 | −1.38 | 0.91 |
| METUNv1_520011 | METUNv2_350013 | Succinate dehydrogenase cytochrome b556 subunit | 324.52 | 191.00 | −1.70 | 0.70 |
| METUNv1_520012 | METUNv2_350014 | Succinate dehydrogenase anchor subunit | 572.13 | 412.89 | −1.39 | 0.88 |
| METUNv1_520013 | METUNv2_350015 | Succinate dehydrogenase flavoprotein subunit | 4086.04 | 2492.77 | −1.64 | 0.84 |
| METUNv1_520014 | METUNv2_350016 | Succinate dehydrogenase Fe–S protein | 843.00 | 709.16 | −1.19 | 0.93 |
| METUNv1_520016 | METUNv2_350018 | Citrate synthase | 2249.29 | 2218.66 | −1.01 | 0.91 |
| METUNv1_520017 | METUNv2_350019 | E1 component of alpha-ketoglutarate dehydrogenase | 1429.24 | 1533.03 | 1.07 | 0.89 |
| METUNv1_520018 | METUNv2_350020 | E2 component of alpha-ketoglutarate dehydrogenase | 697.75 | 762.99 | 1.09 | 0.90 |
| METUNv1_520019 | METUNv2_350021 | E3 component of alpha-ketoglutarate dehydrogenase | 1364.02 | 1401.04 | 1.03 | 0.89 |
| METUNv1_620012 | METUNv2_450011 | Isocitrate dehydrogenase kinase/phosphatase | 241.63 | 308.13 | 1.28 | 0.84 |
| METUNv1_620013 | METUNv2_450012 | Isocitrate dehydrogenase (NADP+) | 2578.02 | 2899.29 | 1.12 | 0.80 |
| Amino Acid Synthesis | ||||||
| METUNv1_660052 | METUNv2_480054 | Glutamate synthase (NADPH) large chain (NADPH-GOGAT) | 3985.94 | 2866.80 | −1.39 | 0.87 |
| METUNv1_660053 | METUNv2_480055 | Glutamate synthase (NADPH) small chain (NADPH-GOGAT) | 1357.89 | 903.01 | −1.50 | 0.91 |
| METUNv1_450044 | METUNv2_280044 | Glutamine synthetase (Glutamate-ammonia ligase) | 2361.78 | 2288.13 | −1.03 | 0.56 |
| METUNv1_470103 | METUNv2_300104 | Glutamate dehydrogenase, NADP-specific (NADP-GDH) | 1040.15 | 1519.76 | 1.46 | 0.93 |
| METUNv1_470104 | METUNv2_300105 | Aspartate aminotransferase (Transaminase A) (AspAT) | 1020.04 | 941.84 | −1.08 | 0.92 |
| METUNv1_470184 | METUNv2_300183 | Putative aspartate transaminase | 617.88 | 620.11 | 1.00 | 0.93 |
| METUNv1_750043 | METUNv2_570043 | Serine-pyruvate aminotransferase | 679.01 | 606.13 | −1.12 | 0.91 |
| Oxidative Phosphorylation | ||||||
| METUNv1_750182 | METUNv2_570182 | ATP synthase F0, A chain | 886.37 | 965.57 | 1.09 | 0.83 |
| METUNv1_750183 | METUNv2_570183 | ATP synthase F0, C chain | 1218.52 | 1365.89 | 1.12 | 0.93 |
| METUNv1_750184 | METUNv2_570184 | ATP synthase F0, B chain | 2528.92 | 2775.88 | 1.10 | 0.89 |
| METUNv1_750185 | METUNv2_570185 | ATP synthase delta chain | 2885.17 | 2564.77 | −1.12 | 0.86 |
| METUNv1_750186 | METUNv2_570186 | ATP synthase subunit alpha subunit | 8915.44 | 6780.46 | −1.31 | 0.89 |
| METUNv1_750187 | METUNv2_570187 | ATP synthase gamma subunit | 5294.58 | 4705.58 | −1.13 | 0.93 |
| METUNv1_750188 | METUNv2_570188 | ATP synthase beta subunit | 9593.01 | 6768.24 | −1.42 | 0.92 |
| METUNv1_750189 | METUNv2_570189 | ATP synthase epsilon subunit | 1784.40 | 1464.79 | −1.22 | 0.90 |
| METUNv1_770336 | METUNv2_590337 | NAD(P) transhydrogenase subunit alpha | 858.91 | 858.01 | −1.00 | 0.93 |
| METUNv1_770337 | METUNv2_590338 | NAD(P) transhydrogenase subunit beta | 399.38 | 455.25 | 1.14 | 0.92 |
| METUNv1_460126 | METUNv2_290122 | NADH-quinone oxidoreductase chain A | 156.74 | 148.38 | −1.06 | 0.93 |
| METUNv1_460127 | METUNv2_290123 | NADH-quinone oxidoreductase subunit B | 451.50 | 376.26 | −1.20 | 0.94 |
| METUNv1_460128 | METUNv2_290124 | NADH (or F420H2) dehydrogenase subunit C | 388.51 | 353.76 | −1.10 | 0.94 |
| METUNv1_460129 | METUNv2_290125 | NADH-ubiquinone oxidoreductase D subunit | 754.27 | 681.62 | −1.11 | 0.92 |
| METUNv1_460130 | METUNv2_290126 | NADH-quinone oxidoreductase subunit E | 207.95 | 211.51 | 1.02 | 0.92 |
| METUNv1_460131 | METUNv2_290127 | NADH-quinone oxidoreductase subunit F | 628.76 | 528.76 | −1.19 | 0.87 |
| METUNv1_460132 | METUNv2_290128 | NADH-quinone oxidoreductase subunit G | 1248.13 | 938.27 | −1.33 | 0.93 |
| METUNv1_460133 | METUNv2_290129 | NADH-quinone oxidoreductase subunit H | 520.13 | 381.75 | −1.36 | 0.93 |
| METUNv1_460134 | METUNv2_290130 | NADH-quinone oxidoreductase subunit I | 327.57 | 337.82 | 1.03 | 0.94 |
| METUNv1_460135 | METUNv2_290131 | NADH-quinone oxidoreductase subunit J | 131.50 | 118.13 | −1.11 | 0.88 |
| METUNv1_460136 | METUNv2_290132 | NADH-quinone oxidoreductase subunit K | 58.88 | 60.63 | 1.03 | 0.91 |
| METUNv1_460137 | METUNv2_290133 | NADH-quinone oxidoreductase subunit L | 637.25 | 465.50 | −1.37 | 0.83 |
| METUNv1_460138 | METUNv2_290134 | NADH-quinone oxidoreductase subunit M | 372.75 | 302.75 | −1.23 | 0.77 |
| METUNv1_460139 | METUNv2_290135 | NADH-ubiquinone oxidoreductase, chain N | 523.88 | 339.07 | −1.55 | 0.64 |
| METUNv1_660132 | METUNv2_480138 | Ferredoxin-NADP reductase | 522.14 | 879.52 | 1.68 | 0.69 |
| METUNv1_590030 | METUNv2_420029 | Ubiquinol-cytochrome c reductase complex, cytochrome c1 | 2503.55 | 1092.90 | −2.29 | 0.58 |
| METUNv1_590031 | METUNv2_420030 | Ubiquinol-cytochrome c reductase complex, cytochrome b | 2945.38 | 1365.76 | −2.16 | 0.81 |
| METUNv1_590032 | METUNv2_420032 | Ubiquinol-cytochrome c reductase iron-sulfur subunit (Rieske iron-sulfur protein) (RISP) | 3125.54 | 1240.80 | −2.52 | 0.84 |
| METUNv1_590008 | METUNv2_420008 | Peroxidase/catalase (Catalase-peroxidase) | 3075.82 | 1883.61 | −1.63 | 0.84 |
| METUNv1_670031 | METUNv2_490032 | Cytochrome c oxidase subunit 1 | 5577.29 | 4801.32 | −1.16 | 0.93 |
| METUNv1_670030 | METUNv2_490031 | Cytochrome c oxidase subunit 2 | 4105.55 | 3835.175 | −1.07 | 0.87 |
| METUNv1_670034 | METUNv2_490035 | Cytochrome c oxidase subunit 3 | 2746.54 | 2227.3 | −1.23 | 0.89 |
| METUNv1_670033 | METUNv2_490034 | Cytochrome c oxidase assembly protein | 1365.325 | 1525.035 | 1.12 | 0.93 |
| METUNv1_580051 | METUNv2_410050 | Hemin uptake protein hemP (fragment) | 1319.89 | 1517.79 | 1.15 | 0.68 |
| METUNv1_580052 | METUNv2_410051 | Bacterioferritin-associated ferredoxin | 930.37 | 739.50 | −1.26 | 0.80 |
| METUNv1_580053 | METUNv2_410052 | Bfr, Bacterioferritin | 1845.02 | 2568.53 | 1.39 | 0.77 |
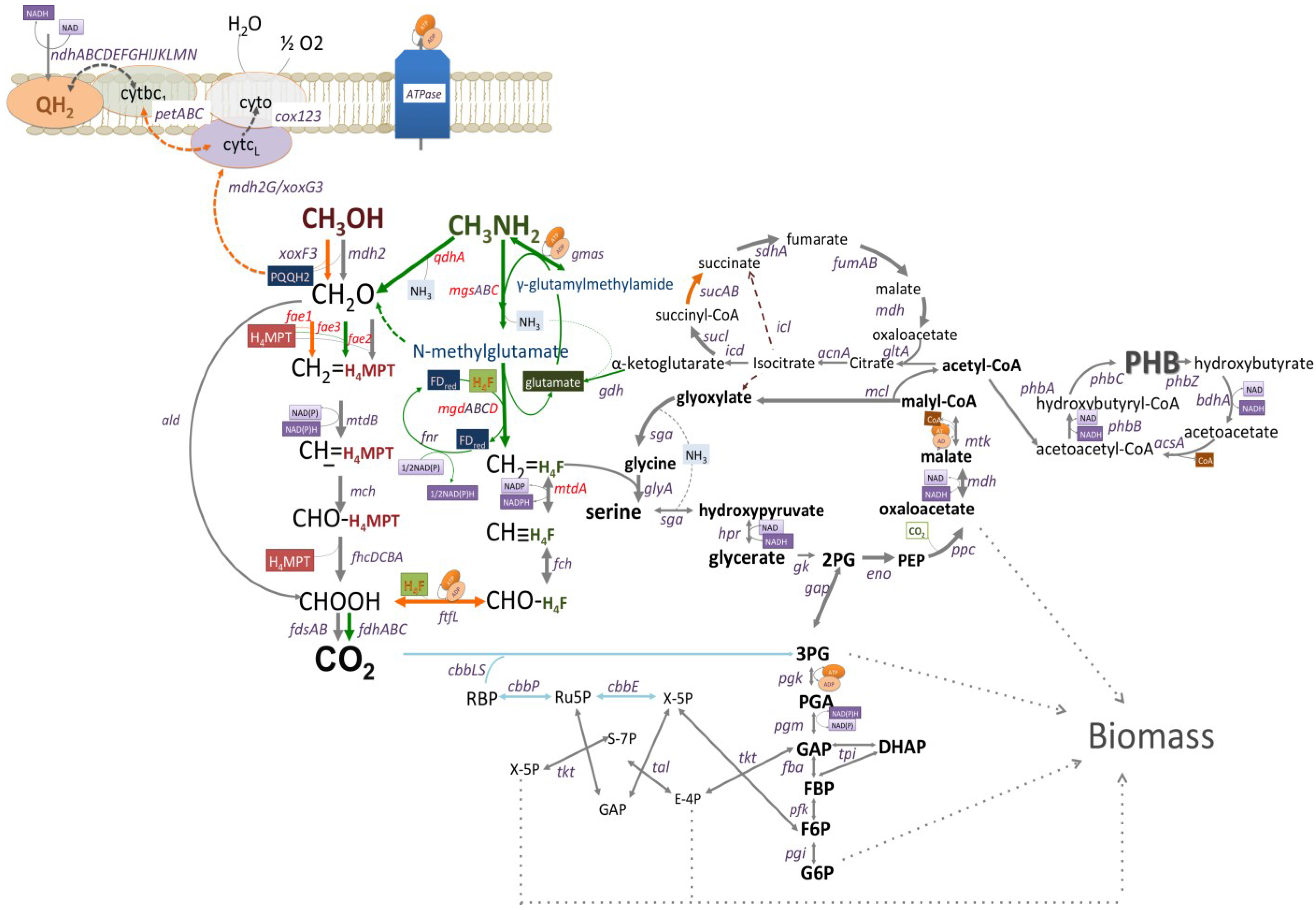
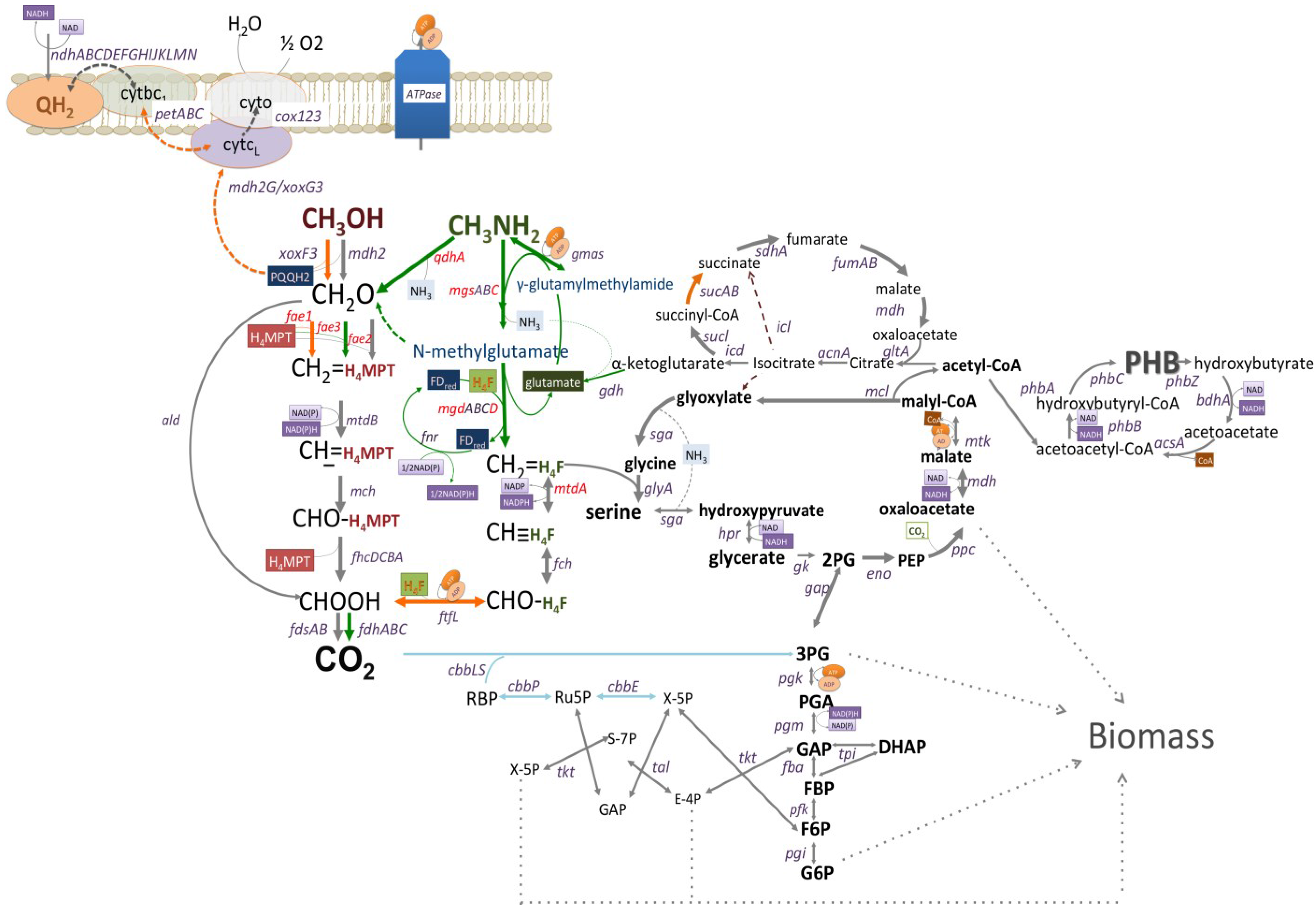
3.2. Mutagenesis Studies: Methylamine Oxidation
3.3. Mutagenesis Studies: Metabolic Arrangement Downstream from the Methylamine Oxidation
| Strain | Enzyme Activity (μmol min−1 mg−1 Protein) |
|---|---|
| FAM5 (WT) | 30 ± 9 |
| ΔqhpA | 6 ± 3 |
4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baytshtok, V.; Kim, S.; Yu, R.; Park, H.; Chandran, K. Molecular and biokinetic characterization of methylotrophic denitrification using nitrate and nitrite as terminal electron acceptors. Water Sci. Technol. 2008, 58, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Qian, L.; Cai, S.; Chen, L. Biodegradation of benazolin-ethyl by strain Methyloversatilis sp. cd-1 isolated from activated sludge. Curr. Microbiol. 2011, 62, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, P.; Alam, S.I.; Bansod, S.; Singh, L. Bacterial diversity of soil samples from the western Himalayas, India. Can. J. Microbiol. 2009, 55, 564–577. [Google Scholar] [CrossRef] [PubMed]
- De Marco, P.; Pacheco, C.C.; Figueiredo, A.R.; Moradas-Ferreira, P. Novel pollutant-resistant methylotrophic bacteria for use in bioremediation. FEMS Microbiol. Lett 2004, 234, 75–80. [Google Scholar]
- Doronina, N.V.; Kaparullina, E.N.; Trotsenko, Y.A. Methyloversatilis thermotolerans sp. nov., a novel thermotolerant facultative methylotroph isolated from a hot spring. Int. J. Syst. Evol. Microbiol. 2014, 64, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Kalyuzhnaya, M.G.; de Marco, P.; Bowerman, S.; Pacheco, C.C.; Lara, J.C.; Lidstrom, M.E.; Chistoserdova, L. Methyloversatilis universalis gen. nov., sp. nov., a novel taxon within the Betaproteobacteria represented by three methylotrophic isolates. Int. J. Syst. Evol. Microbiol. 2006, 56, 2517–2522. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Padron, E.; Bordenave, S.; Lin, S.; Bhaskar, I.M.; Dong, X.; Sensen, C.W.; Fournier, J.; Voordouw, G.; Gieg, L.M. Carbon and sulfur cycling by microbial communities in a gypsum-treated oil sands tailings pond. Environ. Sci. Technol. 2011, 45, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Kalyuzhnaya, M.G.; Hristova, K.; Lidstrom, M.E.; Chistoserdova, L. Characterization of a novel methanol dehydrogenase in representatives of Burkholderiales: Implications for environmental detection of methylotrophy and evidence for convergent evolution. J. Bacteriol. 2008, 190, 3817–3823. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, C.H.; Hristova, K.; Hanada, S.; Meng, X.Y.; Hanson, J.R.; Scow, K.M.; Kamagata, Y. Methylibium petroleiphilum gen. nov., sp. nov., a novel methyl tert-butyl ether-degrading methylotroph of the Betaproteobacteria. Int. J. Syst. Evol. Microbiol. 2006, 56, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Smalley, N.E.; Taipale, S.; de Marco, P.; Doronina, N.V.; Kyrpides, N.; Shapiro, N.; Woyke, T.; Kalyuzhnaya, M.G. Functional and genomic diversity of methylotrophic Rhodocyclaceae: description of the new species Methyloversatilis discipulorum sp. nov. Int. J. Syst. Evol. Microbiol 2015, in press. [Google Scholar]
- Hristova, K.R.; Schmidt, R.; Chakicherla, A.Y.; Legler, T.C.; Wu, J.; Chain, P.S.; Scow, K.M.; Kane, S.R. Comparative transcriptome analysis of Methylibium petroleiphilum PM1 exposed to the fuel oxygenates methyl tert-butyl ether and ethanol. Appl. Environ. Microbiol. 2007, 73, 7347–7357. [Google Scholar] [CrossRef] [PubMed]
- Latypova, E.; Yang, S.; Wang, Y.S.; Wang, T.; Chavkin, T.A.; Hackett, M.; Schäfer, H.; Kalyuzhnaya, M.G. Genetics of the glutamate-mediated methylamine utilization pathway in the facultative methylotrophic beta-proteobacterium Methyloversatilis universalis FAM5. Mol. Microbiol. 2010, 75, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Kalyuzhnaya, M.G.; Chandran, K. Comparative proteomic analysis reveals insights into anoxic growth of Methyloversatilis universalis FAM5 on methanol and ethanol. Environ. Microbiol. 2012, 14, 2935–2945. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Metabolism of methylamine in the tea plant (Thea sinensis L.). Biochem. J. 1973, 132, 753–763. [Google Scholar] [PubMed]
- Jones, J.G.; Bellion, E. In vivo 13C and 15N NMR studies of methylamine metabolism in Pseudomonas species. J. Biol. Chem. 1991, 266, 11705–11713. [Google Scholar] [PubMed]
- Nayak, D.D.; Marx, C.J. Methylamine utilization via the N-methylglutamate pathway in Methylobacterium extorquens PA1 involves a novel flow of carbon through C1 assimilation and dissimilation pathways. J. Bacteriol. 2014, 196, 4130–4139. [Google Scholar] [CrossRef] [PubMed]
- Anthony, C. The Biochemistry of Methylotrophs; Academic Press INC: London, UK, 1982; p. 431. [Google Scholar]
- Chen, Y.; McAleer, K.L.; Murrell, J.C. Monomethylamine as a nitrogen source for a nonmethylotrophic bacterium, Agrobacterium tumefaciens. Appl. Environ. Microbiol. 2010, 76, 4102–4104. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Scanlan, J.; Song, L.; Crombie, A.; Rahman, M.T.; Schäfer, H.; Murrell, J.C. γ-Glutamylmethylamide is an essential intermediate in the metabolism of methylamine by Methylocella silvestris. Appl. Environ. Microbiol. 2010, 76, 4530–4537. [Google Scholar] [CrossRef] [PubMed]
- Gruffaz, C.; Muller, E.E.; Louhichi-Jelail, Y.; Nelli, Y.R.; Guichard, G.; Bringel, F. Genes of the N-methylglutamate pathway are essential for growth of Methylobacterium extorquens DM4 with monomethylamine. Appl. Environ. Microbiol. 2014, 80, 3541–3550. [Google Scholar] [CrossRef] [PubMed]
- Wischer, D.; Kumaresan, D.; Johnston, A.; Khawand, M.E.L.; Stephenson, J.; Hillebrand-Voiculescu, A.M.; Chen, Y.; Murrell, C.J. Bacterial metabolism of methylated amines and identification of novel methylotrophs in Movile Cave. ISME J. 2015, 9, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.A.C.; McTaggart, T.L.; Setboonsarng, U.; Vorobev, A.; Goodwin, L.; Shapiro, N.; Woyke, T.; Kalyuzhnaya, M.G.; Lidstrom, M.E.; Chistoserdovaand, L. Multiphyletic origins of methylotrophy in Alphaproteobacteria, exemplified by comparative genomics of Lake Washington isolates. Environ. Microbiol. 2015, 17, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Kittichotirat, W.; Good, N.M.; Hall, R.; Bringel, F.; Lajus, A.; Médigue, C.; Smalley, N.E.; Beck, D.; Bumgarner, R.; Vuilleumier, S.; et al. Genome sequence of Methyloversatilis universalis FAM5T, a methylotrophic representative of the order Rhodocyclales. J. Bacteriol. 2011, 193, 4541–4542. [Google Scholar] [CrossRef] [PubMed]
- Durham, D.R.; Perry, J.J. Purification and characterization of a heme-containing amine dehydrogenase from Pseudomonas putida. J. Bacteriol. 1978, 134, 837–843. [Google Scholar] [PubMed]
- Nakai, T.; Deguchi, T.; Frébort, I.; Tanizawa, K.; Okajima, T. Identification of genes essential for the biogenesis of quinohemoprotein amine dehydrogenase. Biochemistry 2014, 53, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Takagi, K.; Torimura, M.; Kawaguchi, K.; Kano, K.; Ikeda, T. Biochemical and electrochemical characterization of quinohemoprotein amine dehydrogenase from Paracoccus denitrificans. Biochemistry 1999, 38, 6935–6942. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Lidstrom, M.E. Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. BioTechniques 2002, 33, 1062–1067. [Google Scholar] [PubMed]
- Simon, R.; Priefer, U.; Pühler, A. A broad host range mobilization system for in vivo genetic engineering: Transposon mutagenesis in Gram-negative bacteria. Nat Biotechnol 1984, 1, 784–791. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1989; p. 1626. [Google Scholar]
- Matsen, J.B.; Yang, S.; Stein, L.Y.; Beck, D.; Kalyuzhnaya, M.G. Global molecular analyses of methane metabolism in methanotrophic Alphaproteobacterium, Methylosinus trichosporium OB3b. Part I. Transcriptomic study. Front. Microbiol. 2013, 7, 1–13. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, T.P.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. PNAS 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.J.; Kosály, G.; Lidstrom, M.E. Formate as the main branch point for methylotrophic metabolism in Methylobacterium extorquens AM1. J. Bacteriol. 2008, 190, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Hagemeier, C.H.; Chistoserdova, L.; Lidstrom, M.E.; Thauer, R.K.; Vorholt, J.A. Characterization of a second methylene tetrahydromethanopterin dehydrogenase from Methylobacterium extorquens AM1. Eur. J. Biochem. 2000, 267, 3762–3769. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L. Modularity of methylotrophy, revisited. Environ. Microbiol. 2011, 13, 2601–2622. [Google Scholar] [CrossRef]
- Chistoserdova, L.; Kalyuzhnaya, M.G.; Lidstrom, M.E. The expanding world of methylotrophic metabolism. Annu. Rev. Microbiol. 2009, 63, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Anthony, C. Methanol oxidation and growth yields in methylotrophic bacteria: A review. Acta Biotechnol. 1983, 3, 261–268. [Google Scholar] [CrossRef]
- Elbehti, A.; Brasseur, G.; Lemesle-Meunier, D. First evidence for existence of an uphill electron transfer through the bc1 and NADH-Q oxidoreductase complexes of the acidophilic obligate chemolithotrophic ferrous ion-oxidizing bacterium Thiobacillus ferrooxidans. J. Bacteriol. 2000, 182, 3602–3606. [Google Scholar] [CrossRef] [PubMed]
- Osyczka, A.; Moser, C.C.; Daldal, F.; Dutton, P.L. Reversible redox energy coupling in electron transfer chains. Nature 2004, 427, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Sievert, S.M.; Scott, K.M.; Klotz, M.G.; Chain, P.S.; Hauser, L.J.; Hemp, J.; Hügler, M.; Land, M.; Lapidus, A.; Larimer, F.W.; et al. Genome of the epsilonproteobacterial chemolithoautotroph Sulfurimonas denitrificans. Appl. Environ. Microbiol. 2008, 74, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Good, N.M.; Lamb, A.; Beck, D.A.C.; Martinez-Gomez, N.C.; Kalyuzhnaya, M.G. C1-Pathways in Methyloversatilis universalis FAM5: Genome Wide Gene Expression and Mutagenesis Studies. Microorganisms 2015, 3, 175-197. https://doi.org/10.3390/microorganisms3020175
Good NM, Lamb A, Beck DAC, Martinez-Gomez NC, Kalyuzhnaya MG. C1-Pathways in Methyloversatilis universalis FAM5: Genome Wide Gene Expression and Mutagenesis Studies. Microorganisms. 2015; 3(2):175-197. https://doi.org/10.3390/microorganisms3020175
Chicago/Turabian StyleGood, Nathan M., Andrew Lamb, David A. C. Beck, N. Cecilia Martinez-Gomez, and Marina G. Kalyuzhnaya. 2015. "C1-Pathways in Methyloversatilis universalis FAM5: Genome Wide Gene Expression and Mutagenesis Studies" Microorganisms 3, no. 2: 175-197. https://doi.org/10.3390/microorganisms3020175
APA StyleGood, N. M., Lamb, A., Beck, D. A. C., Martinez-Gomez, N. C., & Kalyuzhnaya, M. G. (2015). C1-Pathways in Methyloversatilis universalis FAM5: Genome Wide Gene Expression and Mutagenesis Studies. Microorganisms, 3(2), 175-197. https://doi.org/10.3390/microorganisms3020175