Abstract
Bacterial persisters are dormant cells that survive antibiotic treatment, serving as a reservoir for the emergence of resistant mutations. The evolution of antibiotic resistance poses a significant challenge to public health. In this study, we investigated the development of resistance in Pseudomonas aeruginosa persister cells by exposing the reference strain PA14 to meropenem and tracked the emergence of resistance mutations over serial passages. Whole-genome sequencing of the populations or individual resistant strains revealed evolutionary trajectories. In the initial passages, low-level meropenem-resistant mutants harbored various mutations, accompanied by increasing population survival. Then, mutations in the oprD gene appeared, followed by mutation in the mexR gene in most of the cells, leading to high-level meropenem resistance and collateral resistance to ciprofloxacin. Our study provides insights into the evolutionary pathways of P. aeruginosa under lethal antibiotic pressure, highlighting the dynamic interplay between persister cells and the emergence of resistance mutations.
1. Introduction
Antibiotic resistance has emerged as one of the most pressing global public health challenges. A large proportion of resistant infections are caused by a group of bacterial pathogens including Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp., which are called ESKAPE [1]. These organisms possess a range of intrinsic and acquired resistance mechanisms that enable them to escape the effects of antimicrobial agents. The persistent use of antibiotics has driven the emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains, rendering many conventional antibiotics ineffective [2]. ESKAPE pathogens are associated with high mortality rates and significantly increased healthcare costs. Among ESKAPE, P. aeruginosa is an opportunistic human pathogen that can cause life-threatening acute and chronic infections [3]. Carbapenems have been considered the most active and potent agents against MDR Gram-negative pathogens [4]. However, efflux pump upregulation, porin mutations, and β-lactamase production, etc., increase bacterial resistance to carbapenems [5,6,7]. Carbapenem-resistant P. aeruginosa (CRPA) is disseminated globally and constitutes a significant threat to public health [8,9]. It is in the “critical” category of the World Health Organization (WHO)’s priority list of bacterial pathogens, for which research and development of new antibiotics is urgently required [10].
P. aeruginosa is a leading cause of nosocomial infections, with carbapenem resistance posing a critical therapeutic challenge [8,9]. Central to this resistance are two chromosomal mechanisms: loss of the OprD porin and hyperactivation of RND efflux pumps via mexR mutations. The OprD porin facilitates carbapenem uptake, and its inactivation—through frameshift mutations, premature stop codons, or large-fragment deletions—reduces membrane permeability, elevating imipenem or meropenum MICs by 4–32-fold. Notably, 79.1% of carbapenem-resistant clinical isolates show oprD downregulation, while 20.9% exhibit overexpression of efflux pumps or carbapenemases [11]. Concurrently, mexR mutations derepress the MexAB-OprM efflux pump, a major transporter of diverse antibiotics, including β-lactams, fluoroquinolones, aminoglycosides, etc. Structural studies reveal that mexR mutations or oxidative-stress-induced disulfide bond formation trigger conformational changes in MexR’s DNA-binding domain, abolishing its repressor function and leading to mexAB-oprM over expression [12]. It has been found that mexR mutants play major roles in antibiotic evolution and drive collateral resistance. Clinically, synergistic oprD loss and MexAB-OprM hyperactivation create a “dual-hit” resistance mechanism, observed in 89% of carbapenem-resistant strains [13].
Bacterial evolution, driven by mechanisms such as mutation and horizontal gene transfer, is a key factor in the development of antimicrobial resistance [14,15,16]. Bacterial persisters are a subpopulation of dormant cells that survive lethal antibiotic treatment through mechanisms such as toxin–antitoxin (TA) systems and metabolic dormancy, leading to chronic and recurrent infections [17,18,19,20]. P. aeruginosa utilizes toxin–antitoxin (TA) systems as critical regulators for stress adaptation, persistence formation, and virulence expression. TA systems typically comprise a stable toxin that disrupts essential cellular processes and a labile antitoxin that neutralizes toxicity. Recent genomic analyses reveal that P. aeruginosa harbors 12–15 type II TA systems across major strains such as PA14 (13 systems), PAO1 (12 systems), and ATCC 27853 (15 systems), with significant variation in mobile genetic elements [21,22,23]. TA systems further drive metabolic reprogramming for persistence. The PA14_51010 proteins in the PA14 strain reduce intracellular NAD+ levels to induce metabolic dormancy and antibiotic tolerance, while the ParE toxin inhibit DNA gyrase to halt replication under nutrient stress. This persistence phenotype facilitates chronic infections, as seen in cystic fibrosis lungs where TA systems coordinate long-term colonization via (p)ppGpp signaling networks [24,25]. In addition, persister cells may serve as a reservoir for the development of resistance mutations over time, as their survival under antibiotic pressure provides an opportunity for mutations to arise [26,27]. Understanding the interplay between persister formation and the emergence of resistance mutations is crucial for developing novel therapeutic strategies to combat persistent infections and prevent the evolution and spread of antibiotic resistance.
In studies of bacterial resistance evolution, in vitro serial passaging experiments have been widely used, in which bacteria are grown in increasing concentrations of antibiotics to select resistant mutants [28,29]. However, resistant mutants arising from non-growing persister are likely to be neglected in the assay. Our hypothesis was that the distinctive physiological status and gene expression profile of persisters might affect the evolution trajectory of resistance. Thus, in this study, we examined the resistance development of P. aeruginosa persister cells following treatment with a lethal dosage of meropenem and characterized the mutation sites as well as collateral resistance.
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, and Primers
The bacterial strains, plasmids, and primers used in this study are detailed in Supplementary Table S1. P. aeruginosa PA14 (ATCC 27853) served as the primary wild-type strain. All strains were cultured aerobically in Luria–Bertani (LB) medium (1% tryptone, 0.5% yeast extract, 1% NaCl, pH 7.4 ± 0.2) at 37 °C with constant shaking at 200 rpm. Long-term storage utilized glycerol stocks (25% v/v) prepared from mid-log-phase cultures (OD600 = 0.8–1.0) and stored at −80 °C, with working strains subcultured on LB agar plates for no more than two passages to minimize genetic drift.
2.2. Antimicrobial Susceptibility Test
Minimum inhibitory concentrations (MICs) of meropenem, ciprofloxacin, and tobramycin were determined by the broth microdilution method in cation-adjusted Mueller–Hinton broth (CAMHB, Oxoid CM0405) following Clinical and Laboratory Standards Institute (CLSI) M100-Ed34 (2024) guidelines [30]. Mid-log-phase cultures were adjusted to 0.5 McFarland standard (~1 × 108 CFU/mL) in sterile saline using a Densimat densitometer (BioMérieux, Marcy-l’Étoile, France) and then diluted 1:100 in CAMHB to achieve a final inoculum of 5 × 105 CFU/well in 96-well microtiter plates. Two-fold serial dilutions of antibiotics (meropenem: 0.015–256 μg/mL; ciprofloxacin: 0.008–32 μg/mL; tobramycin: 0.25–128 μg/mL) were prepared using an electronic multichannel pipette (Eppendorf (Hamburg, Germany) EpMotion 5075t) to ensure volumetric accuracy. Each plate included P. aeruginosa ATCC 27853 as quality control strains, with the MIC defined as the lowest concentration showing ≥90% growth inhibition after 18 ± 2 h of incubation at 35 ± 2 °C under visual inspection.
2.3. Bacteria Killing Assay
Briefly, overnight bacterial cultures were diluted into fresh LB to an OD600 of 0.05. The bacteria were cultured for 2 h until the OD600 reached approximately 0.8~1.0. Then, the bacteria were treated with 8 μg/mL meropenem. At each indicated time point, an aliquot of the cultures was taken out. The bacteria were washed with 0.9% NaCl three times, followed by serial dilution and plating on LB agar for colony-forming unit (CFU) counting [31,32]. All experiments were conducted in biological triplicates.
2.4. Experimental Evolution of Persister Cells
The overnight culture of wild-type PA14 was diluted 1:100 in 3 mL of fresh LB. The bacteria were grown at 37 °C, 200 rpm until the OD600 reached 0.8–1.0, followed by treatment with 8 μg/mL meropenem for 6 h. A total of 100 μL of the bacteria was used to determine the number of live bacteria by plating. The remaining cells were subjected to centrifugation in 0.3 M sucrose at 12,000 rpm to remove dead cell debris. The bacteria were then resuspended in 3 mL of fresh LB and culture to an OD600 of 1.0. A total of 100 μL of the bacteria was plated on plates containing indicated antibiotics (meropenem, ciprofloxacin, and tobramycin) at different concentrations to determine resistance mutation rates. The remaining bacteria were subjected to the next round of meropenem (8 μg/mL) treatment. This process was repeated until 8 μg/mL of meropenem was no longer capable of killing the bacteria. For the control group, cultures were grown under the same conditions (37 °C, 200 rpm) until the OD600 reached 0.8–1.0. Similarly, 100 μL of the bacteria was plated on plates containing the indicated antibiotics at different concentrations. The remaining bacteria were processed in accordance with the experimental group and subjected to the same number of rounds as the experimental group.
2.5. Genomic Sequencing
Individual clones were subjected to next-generation whole-genome sequencing conducted by Shanghai Honsun Biological Technology Company. Whole-genome sequencing with a depth of 200× of the heterogenous bacterial pools was conducted by the Tianjin Uniteomics Company (Tianjin, China). Briefly, bacterial genomic DNA was extracted with a DNA purification kit (Tiangen Biotech, Beijing, China). Fragments smaller than 500 bp were obtained from 200 ng genomic DNA by sonication (Covaris S220, Covaris, Woburn, MA, USA), followed by end treatment and adaptor ligation. Adaptor-ligated DNA fragments of about 470 bp were recovered and then PCR-amplified for six cycles, and the PCR products were cleaned up, validated using a Qsep100 (Bioptic, Taiwan, China), and quantified by a Qubit3.0 Fluorometer (Invitrogen, Carlsbad, CA, USA). Sequencing was carried out using a 2 × 150 paired-end (PE) configuration on an Illumina Hiseq instrument according to manufacturer’s instructions (Illumina, San Diego, CA, USA). The data were aligned to the PA14 reference genome (NC_002516.2) via the BW2 software (version 0.7.12). Single-nucleotide variation (SNV) or InDel mutation were detected using the software Samtools (version 1.1) and the Unified Genotyper module from GATK (version 4.5.0.0).
3. Results
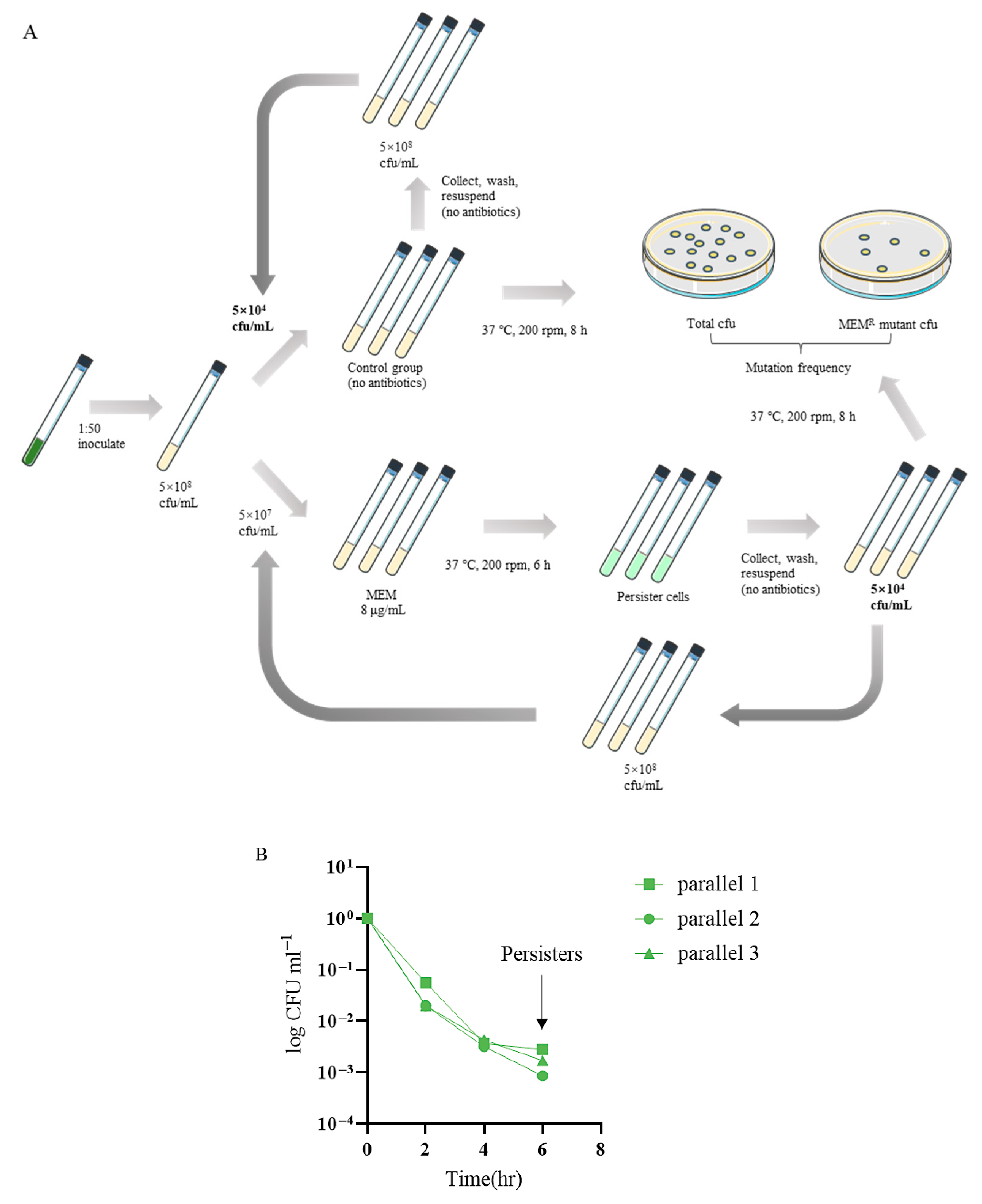
To investigate the dynamics and evolutionary trajectories of resistance developed from persister cells, we serially passaged the P. aeruginosa reference strain PA14 in a meropenem killing assay (Figure 1A). Three parallels of the bacteria were treated with 8 μg/mL meropenem (clinical break point), resulting in a typical biphasic killing curve (Figure 1B). At 6 h following the treatment, the bacteria were killed at a slow rate, representing a persister population (Figure 1B). The live bacteria were collected and grown in a fresh medium to an OD600 of 1.0, which was subjected to next round of meropenem killing. The dynamics of bacterial survival and antibiotic resistance frequencies were determined.
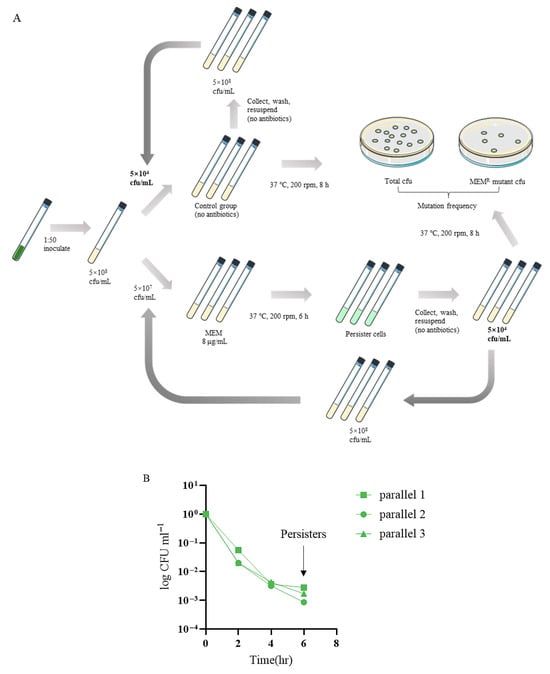
Figure 1.
(A) The experimental flow chart. (B) The typical biphasic killing curve.
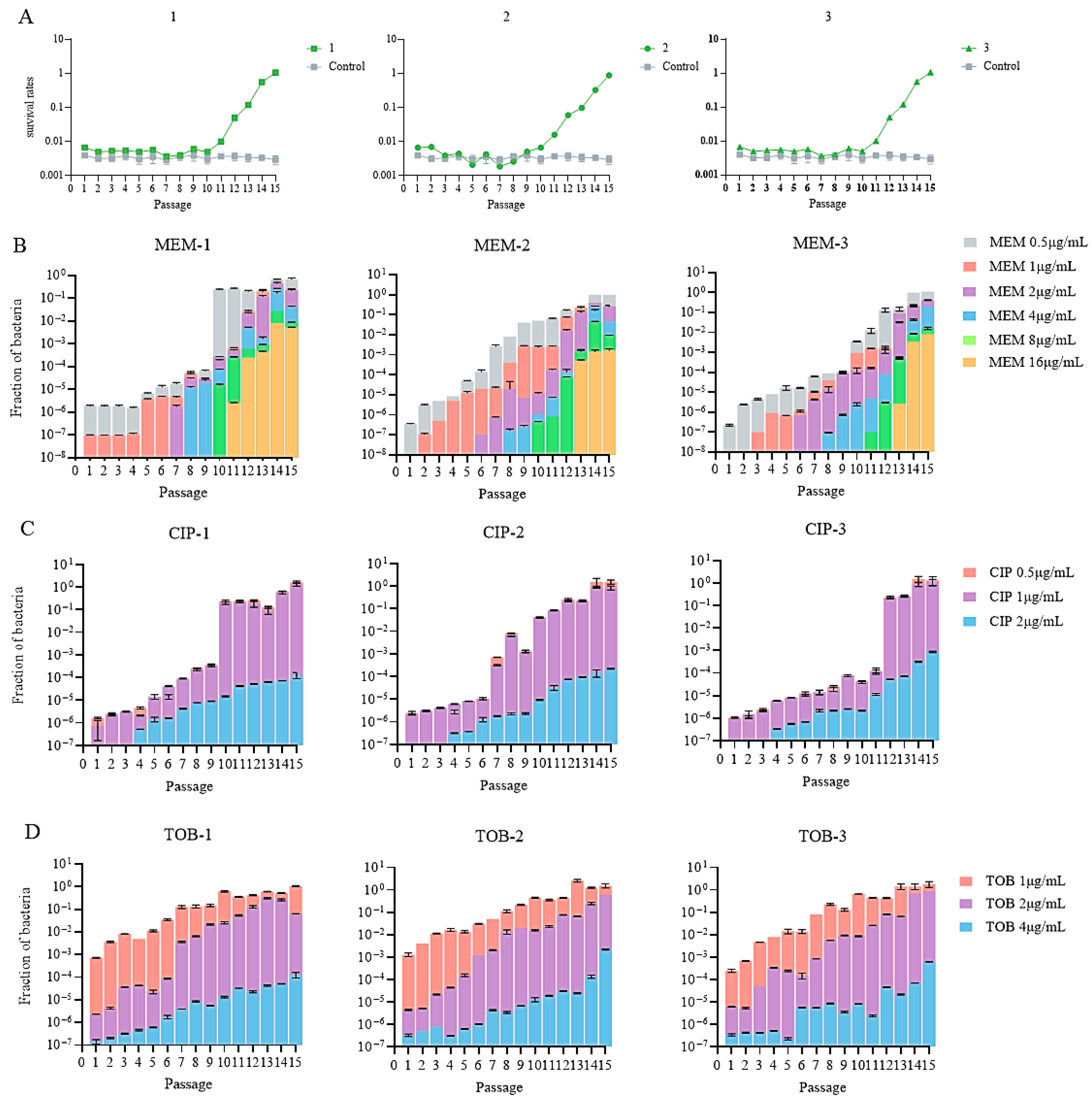
In the first 9–10 passages, the survival rates of the meropenem-treated groups remained at around 0.3%, followed by a quick increase to 100% over the next 5–6 passages (Figure 2A). In contrast, no increase in survival was observed in the cells passaged in antibiotic-free medium (Figure 2A). To determine the development of meropenem and collateral resistance after each passage, the recovered bacteria were plated on plates in the absence or presence of increasing amounts of meropenem, ciprofloxacin, and tobramycin. Clinically defined meropenem-resistant bacteria (MIC ≥ 16 μg/mL) arose at the 11th or 13th passage, in which the corresponding survival rates were 1% or 10% (Figure 2A,B). By 15 passages, around 1% of the bacteria were able to form colonies on plates containing 16 μg/mL meropenem (Figure 2B). Meanwhile, at least 0.01% of the bacteria were able to form colonies on plates containing ciprofloxacin and tobramycin at the concentrations of the clinical breakpoints (2 and 4 μg/mL, respectively) (Figure 2C,D).
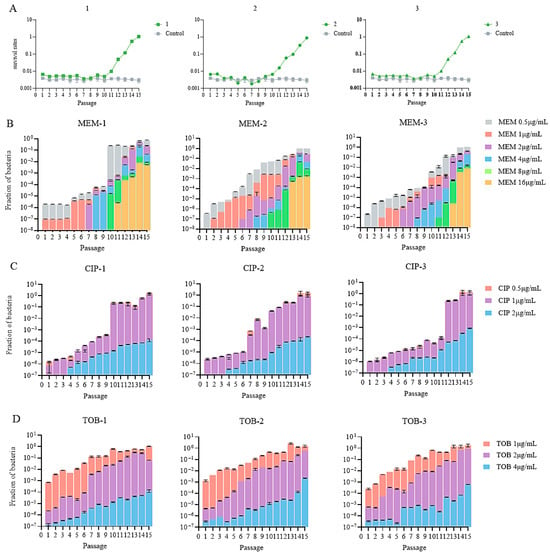
Figure 2.
(A) Survival rates in the three parallels. (B–D) Mutation rates of the bacterial population after meropenem treatment against meropenem (MEM), ciprofloxacin (CIP), and tobramycin (TOB).
To identify mutations associated with the development of resistance in these populations, the heterogenous bacterial pools in the final passage were subjected to whole-genome sequencing with a depth of 200×. Variations were identified at a 5% cut-off. A mexR T130P mutation was identified in all the three parallel populations with percentages of 100%, 99.55%, and 100%. Additionally, the proportions of oprD mutations in the populations were 41.36%, 69.35%, and 40.97%, respectively. We then isolated three single colonies on the 16 μg/mL meropenem plates from each of the parallels. The MICs of meropenem for the isolates reached 64 μg/mL. Meanwhile, the MICs of ciprofloxacin and tobramycin were both 0.5 μg/mL (Table 1).

Table 1.
The MIC of single colonies on the 16 μg/mL meropenem plates at the 15th passage.
Whole-genome sequencing revealed that all the isolates harbored the mexR T130P mutation with various oprD mutations (Table 2).
Table 2.
The mexR and oprD mutations of the single colonies on the 16 μg/mL meropenem plates at the 15th passage.
To validate the contribution of mexR and oprD gene mutations in antibiotic resistance, we constructed gene deletion mutants, the mexR T130P mutant and complementation strains, followed by antibiotic resistance evaluation (MIC test). Deletion of mexR in wild-type PA14 increased the MIC of meropenem and ciprofloxacin to 4 and 0.5 μg/mL, respectively, whereas the MIC of tobramycin was not affected. Deletion of oprD increased the MIC of meropenem to 8 μg/mL without affecting the MICs of ciprofloxacin and tobramycin. Meanwhile, deletion of both oprD and mexR increased the MIC of meropenem to 64 μg/mL, whereas the MICs of ciprofloxacin and tobramycin remained the same as the ΔmexR mutation (0.5 μg/mL) (Table 3). All the increased MICs were restored by complementation with a mexR gene (Table 3). These results verified that mutation of oprD increases bacterial resistance to meropenem, and mutation of mexR contributes to resistance to meropenem, ciprofloxacin, and tobramycin. In addition, mutations of oprD and mexR synergistically increase bacterial resistance to meropenem.
Table 3.
Bacterial antibiotic resistance levels (MIC) of P. aeruginosa strains.
To explore the evolutionary trajectory, we isolated three single colonies on the 16 μg/mL meropenem plates at the earliest passage with the appearance of colonies from each of the parallels, i.e., passages 11th, 13th, and 13th passage of parallels 1, 2, and 3, respectively (Figure 2B). Whole-genome sequencing revealed mutations in mexR and oprD in all the strains. No mutation in other known resistance-related genes were identified. We then focused on the mutation trajectory of these two genes.
Additionally, we observed that, relative to the reference genome, the lasR gene in the control group contained two mutations, whereas lasR in the experimental groups remained consistent with the wild type.
From each parallel, we isolated six colonies on the plates containing 1, 2, 4, and 8 μg/mL meropenem that appeared at the earliest passages (Table 4). The MICs of the strains were determined (Table 4), and the oprD and mexR genes were sequenced (Table 5).
Table 4.
Bacterial antibiotic resistance levels (MIC) of single colonies isolated from plates containing indicated concentrations of meropenem.
Table 5.
Mutations in the oprD and mexR genes in stains isolated from plates containing indicated concentrations of meropenem.
In all the strains isolated from 8 μg/mL meropenem plates, both the mexR T130P mutation and mutations in the oprD gene were present (Table 5). In the strains isolated from 4 μg/mL meropenem plates, fourteen strains contained mutations in oprD; however, no mexR mutation was identified (Table 5). In all the strains isolated from 2, 1, and 0.5 μg/mL meropenem plates, no mutation in either mexR or oprD was present. To identify mutations that rose before the oprD and mexR mutations, the bacterial population of each parallel at the sixth or seventh passage (the earliest passages with colonies on 2 μg/mL meropenem plates) were subjected to whole-genome sequencing with a depth of 200×.
Mutations in the genes vgrG1b, phzD2, vgrG14, and cdrA were present with percentage of 5.86–100% in all the sequenced populations from the 7th or 6th to the 15th passages (Tables S3–S5). Mutations in 28 genes were detected at a percentage of 5.59–26.73% at the seventh or sixth passages. However, those mutations were not detected in the clones from the 10–15th passages as well as single colonies and the whole population at the 15th passage (Tables S3–S5). Among those 28 mutated genes, sltB1 (allele frequency 5.42%) in Group 1 (Generation 7th) encodes a lytic transglycosylase B that cuts peptidoglycan glycans.
4. Discussion
Bacterial persisters are cells that survived lethal dosages of antibiotics, which contribute to recurrent and chronic infections [33,34]. Meanwhile, these cells provide a reservoir for mutations that contribute to the development of antibiotic resistance [35,36]. The alarmone molecule (p)ppGpp and TA systems play important roles in persister formation [37]. They also affect global gene expression [38]. In addition, how (p)ppGpp, TA systems together with lethal antibiotic pressure affect mutation frequency and evolutionary trajectories remains to be explored. Here, in this study, we determined the resistance evolution and mutations by employing continuous passaging of P. aeruginosa cells under lethal concentrations of meropenem (16× MIC). Traditional investigations into bacterial resistance evolution overwhelmingly utilize sub-inhibitory concentrations (sub-MIC) of antibiotics to select for resistant mutants over serial passages [18,39,40]. While the traditional approach successfully models resistance emergence in environments with sub-inhibitory drug levels, it inherently excludes the evolutionary trajectories possible only under the bactericidal pressure. Our approach deliberately subjects bacterial populations to concentrations where only persisters or variants possessing exceptional immediate tolerance mechanisms can survive, simulating scenarios encountered at peak antibiotic concentrations or within poorly penetrated niches during aggressive chemotherapy. This high-stress environment imposes a dramatically different selective pressure compared to sub-MIC studies [41,42]. This design might provide novel insights into the resilience mechanisms and resistance evolution pathways.
Central to the evolved resistance observed under lethal meropenem stress were mutations in the well-characterized genes mexR and oprD. It has been demonstrated that mutations in the mexR and oprD genes result in upregulation of the multi-drug efflux system MexAB-OprM and a reduction in carbapenem uptake, respectively [43,44,45,46]. Crucially, our findings demonstrate that even under a bactericidal pressure far exceeding the MIC, these “classical” mechanisms retain substantial potency and were the dominant drivers of resistance development. This observation broadens the perceived applicability of these mutations. Prior studies extensively document their importance at sub-MIC or inhibitory concentrations in vitro and in vivo, where incremental increases in resistance enable survival and growth [47,48,49,50]. Our data show that the hyperexpression of the efflux system caused by mexR mutations and the impermeability caused by oprD loss are highly effective survival mechanisms under lethal dosages of meropenem. The persistent dominance of these mutations under lethal selective regimes underscores their important roles in P. aeruginosa carbapenem resistance and evidences their emergence during aggressive high-dose therapy.
In our passaging assays, lasR-null mutants emerged in the control groups, which is consistent with previous reports. In addition, lasR mutants have been identified from patients with chronic P. aeruginosa infections [51,52,53]. Clinically, lasR defects confer multidrug resistance (MDR) via efflux pump hyperactivation (e.g., MexXY/OprM- and NalC-mediated pathways), reducing intracellular antibiotic accumulation of β-lactams and aminoglycosides [54,55,56]. Inactivation of lasR correlates with mutations in efflux regulators (nalC, mexZ) [55,56]. However, lasR loss also imposes fitness costs. Mutants exhibit metabolic reprogramming toward anaerobic respiration via nitrate reductase (nar), enhancing survival in hypoxic biofilm niches but increasing susceptibility to nitrosative stress [57,58]. Crucially, this metabolic vulnerability may drive reversion to wild-type lasR under conditions where redox balance is critical [59,60]. Additionally, lasR mutants display bimodal persistence strategies: they enrich phenazine biosynthesis and EPS production, shielding persisters from immune clearance but compromise quorum-sensing (QS)-coordinated virulence, potentially reducing acute infectivity [58]. The epistatic interaction between lasR and secondary mutations further modulates resistance. As reported, fusA1 mutations enhance MexXY efflux activity, amplifying tobramycin resistance in lasR mutants [59,60]. The absence of the lasR mutation in the meropenum treatment groups indicate that there might be a fitness cost of the lasR mutations under the pressure of a lethal dosage of meropenum. In vivo experiments are warranted to examine the development of lasR mutations in the presence of antibiotic treatment.
Beyond the established mexR/oprD-related mechanisms, genomic sequencing revealed mutations in genes not previously known to directly relate to meropenem resistance in P. aeruginosa. It has been reported that mutation of sltB1 impairs the incorporation of a new peptidoglycan subunit into the cell wall, resembling low-level beta-lactam treatment, which leads to ampC upregulation and subsequent increased resistance to β-lactam antibiotics [61,62,63,64]. While the other genes lack established roles in drug efflux, enzymatic modification of antibiotics, or target alteration, the occurrence of their mutations in the resistant mutants suggests a potentially contributory role. We postulate that these mutations might increase bacterial tolerance, acting as crucial steppingstones under a lethal dosage of meropenem [65,66]. The mechanisms might be an alteration in metabolic flux, membrane composition, stress response pathways (e.g., envelope stress), or persister formation [67,68]. These early “enabler” mutations likely expand the population’s evolutionary opportunities by increasing bacterial survival under the lethal antibiotic stress [69]. This highlights a complex trajectory where initial stress adaptations preceded and potentiate the development of major resistance mutations under severe drug pressure. Functions of the early-stage mutated genes warrants further studies. A previous study in Escherichia coli demonstrated that resistant mutant cells in the bacterial population secrete indole, thereby improving the survival of the whole population [70]. Thus, the appearance of meropenem-resistant mutants might enhance the survival of the population, providing a bigger reservoir for additional mutations.
In addition, our study reveals that the meropenem-resistant mutants developed collateral resistance to ciprofloxacin and tobramycin. This phenomenon is most likely due to the mexR mutation, which results in upregulation of the MexAB-OprM efflux pump [43,44,45,46]. The collateral resistance may impair the treatment efficacies of ciprofloxacin and tobramycin despite no prior exposure [71]. Meanwhile, our data aligns with genomic surveys of XDR P. aeruginosa outbreaks, wherein mexR mutants exhibit 89% co-resistance to carbapenems, fluoroquinolones, and aminoglycosides, which confirmed this evolutionary trajectory’s relevance to clinics [72,73].
The passages in this study were performed in a complete LB medium in vitro. However, in clinical settings, bacteria encounter antibiotics in the host environment. The nutritional condition is significantly different from the in vitro culture medium. Meanwhile, the bacteria are under the pressure of the host’s immune clearance. This environment may alter the bacterial evolutionary trajectory of antibiotic resistance. For instance, mutations that increase antibiotic resistance while incurring fitness costs in the host environment are likely to be lost in vivo. Therefore, passaging assays in mouse infection models may reveal more clinic relevant bacterial evolutionary trajectories of antibiotic resistance.
5. Conclusions
This study delineates a hierarchical evolutionary trajectory in P. aeruginosa persister populations under lethal meropenem stress, which is different from conventional sub-inhibitory resistance evolution. We demonstrate that mexR and oprD mutations remain the dominant drivers of high-level meropenem resistance even under bactericidal pressure, confirming their major roles in efflux hyperactivation and porin-mediated impermeability against meropenem. Crucially, early mutations in engA and sltB1, previously unreported in carbapenem resistance, might function as evolutionary enablers, probably enhancing transient population survival and expanding the genetic reservoir for subsequent high-impact mutations. This adaptive cascade (enabler→oprD loss→mexR-mutation-mediated hyper-resistance) reveals an evolution trajectory under the lethal selective environments, providing a clue for understanding recalcitrant infections during high-dose therapy.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms13071672/s1, Table S1: Bacteria and plasmids used in this study; Table S2: Primers used in this study; Table S3: Group 1 7th-generation mutation genes; Table S4: Group 2 6th-generation mutation genes; Table S5: Group 3 6th-generation mutation genes. Figure S1: Group 1 generation 7th allele mutant frequency trajectories; Figure S2: Group 2 generation 6th allele mutant frequency trajectories; Figure S3: Group 3 generation 6th allele mutant frequency trajectories. References [74,75] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, W.W. and S.J.; methodology, W.W., C.X. and J.F.; validation, Z.C., Y.J. and Y.B.; formal analysis, J.F.; investigation, J.F.; data curation, J.F.; writing—original draft preparation, J.F.; writing—review and editing, W.W.; visualization, J.F.; supervision, W.W. and S.J.; project administration, W.W.; funding acquisition, W.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (32470193, 32170177), Nankai University Tianjin Applied and Fundamental Research Project (22JCZDJC0041), and Natural Science Foundation of Tianjin (21JCZDJC00210, 22JCYBJC00790, 22JCYBJC00560).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original data presented in the study are openly available in the NCBI SRA database at accession number PRJNA1268141.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| MEM | meropenem |
| CIP | ciprofloxacin |
| TOB | tobramycin |
References
- Shortridge, D.; Gales, A.C.; Streit, J.M.; Huband, M.D.; Tsakris, A.; Jones, R.N. Geographic and temporal patterns of antimicrobial resistance in Pseudomonas aeruginosa over 20 years from the SENTRY antimicrobial surveillance program, 1997–2016. Open Forum Infect. Dis. 2019, 6 (Suppl. 1), S63–S68. [Google Scholar] [CrossRef] [PubMed]
- Botelho, J.; Grosso, F.; Peixe, L. Antibiotic resistance Pseudomonas aeruginosa—Mechanisms, epidemiology and evolution. Drug Resist. Updates 2019, 44, 100640. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y. Treatment Options for Carbapenem-resistant Gram-negative Bacterial Infections. Clin. Infect. Dis. 2019, 69 (Suppl. 7), S565–S575. [Google Scholar] [CrossRef] [PubMed]
- Durante-Mangoni, E.; Andini, R.; Zampino, R. Management of carbapenem-resistant Enterobacteriaceae infections. Clin. Microbiol. Infect. 2019, 25, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Sabour, S.; Harrington, K.R.V.; Martinson, E.; Bhatnagar, A.S.; Huang, J.Y.; Duffy, D.; Bantle, K.; Lutgring, J.D.; Karlsson, M.; Brown, A.C. Characterization of carbapenem-resistant Enterobacterales and Pseudomonas aeruginosa carrying multiple carbapenemase genes-Antimicrobial Resistance Laboratory Network, 2018–2022. J. Clin. Microbiol. 2024, 62, e0122024. [Google Scholar] [CrossRef] [PubMed]
- Tenover, F.C.; Nicolau, D.P.; Gill, C.M. Carbapenemase-producing Pseudomonas aeruginosa—An emerging challenge. Emerg. Microbes Infect. 2022, 11, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Karampatakis, T.; Antachopoulos, C.; Tsakris, A.; Roilides, E. Molecular epidemiology of carbapenem-resistant Pseudomonas aeruginosa in an endemic area: Comparison with global data. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Brink, A.J. Epidemiology of carbapenem-resistant Gram-negative infections globally. Curr. Opin. Infect. Dis. 2019, 32, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Ribot, P.A.; Cabot, G.; Mulet, X.; Periañez, L.; Martín-Pena, M.L.; Juan, C.; Pérez, J.L.; Oliver, A. Mechanisms leading to in vivo ceftolozane/tazobactam resistance development during the treatment of infections caused by MDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yi, C.; Zhang, J.; Zhang, W.; Ge, Z.; Yang, C.; He, C. Structural insight into the oxidation-sensing mechanism of the antibiotic resistance of regulator MexR. EMBO Rep. 2010, 11, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.R.; Pitton, M.; Oberhaensli, S.; Schlegel, K.; Prod’hOm, G.; Blanc, D.S.; Jakob, S.M.; Que, Y.-A.; Goldberg, J.B. Parallel Evolution of Pseudomonas aeruginosa during a Prolonged ICU-Infection Outbreak. Microbiol. Spectr. 2022, 10, e0274322. [Google Scholar] [CrossRef] [PubMed]
- Souque, C.; González Ojeda, I.; Baym, M. From Petri Dishes to Patients to Populations: Scales and Evolutionary Mechanisms Driving Antibiotic Resistance. Annu. Rev. Microbiol. 2024, 78, 361–382. [Google Scholar] [CrossRef] [PubMed]
- Nicoloff, H.; Hjort, K.; Andersson, D.I.; Wang, H. Three concurrent mechanisms generate gene copy number variation and transient antibiotic heteroresistance. Nat. Commun. 2024, 15, 3981. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: Our worst nightmare? Clin. Infect. Dis. 2002, 34, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.B.; Sidders, A.E.; Velez, A.Z.; Hanson, B.M.; Angeles-Solano, M.; Ruffin, F.; Rowe, S.E.; Arias, C.A.; Fowler, V.G., Jr.; Thaden, J.T.; et al. In-patient evolution of a high-persister Escherichia coli strain with reduced in vivo antibiotic susceptibility. Proc. Natl. Acad. Sci. USA 2024, 121, e2314514121. [Google Scholar] [CrossRef] [PubMed]
- Levin-Reisman, I.; Ronin, I.; Gefen, O.; Braniss, I.; Shoresh, N.; Balaban, N.Q. Antibiotic tolerance facilitates the evolution of resistance. Science 2017, 355, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q. Persistence: Mechanisms for triggering and enhancing phenotypic variability. Curr. Opin. Genet. Dev. 2011, 21, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Yadav, M.; Singh, G.; Chaudhary, S.; Ghosh, C.; Rathore, J.S. Decoding the TAome and computational insights into parDE toxin-antitoxin systems in Pseudomonas aeruginosa. Arch. Microbiol. 2024, 206, 360. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Liu, J.; Huang, Y.S.; Chen, W.M.; Lin, J. Cyclic Diguanylate G-Quadruplex Inducer-Quorum Sensing Inhibitor Hybrids as Bifunctional Anti-biofilm and Anti-virulence Agents Against Pseudomonas aeruginosa. J. Med. Chem. 2024, 67, 18911–18929. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, H.; Wang, L.; Tao, R.; Song, G.; Cao, L.; Yan, W.; Wu, Z.; Liu, Q.; Chen, Y.; et al. A plasmid-encoded inactive toxin-antitoxin system MtvT/MtvA regulates plasmid conjugative transfer and bacterial virulence in Pseudomonas aeruginosa. Nucleic Acids Res. 2025, 53, gkaf075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, T.; Lu, W.; Lin, Y.; Zhou, M.; Cai, X. Hybrid Cell Membrane-Engineered Nanocarrier for Triple-Action Strategy to Address Pseudomonas aeruginosa Infection. Adv. Sci. 2025, 12, e2411261. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, S.; Zhao, N.; Nong, C.; He, Y.; Bao, R. Pseudomonas aeruginosa two-component system CprRS regulates HigBA expression and bacterial cytotoxicity in response to LL-37 stress. PLoS Pathog. 2024, 20, e1011946. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Wu, Z.; Liang, W.; Zhang, X.; Cai, X.; Li, J.; Liang, L.; Lin, D.; Stoesser, N.; Doi, Y.; et al. Prediction of Antibiotic Resistance Evolution by Growth Measurement of All Proximal Mutants of Beta-Lactamase. Mol. Biol. Evol. 2022, 39, msac086. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.L.; Zhang, S.; Wang, Z.; Song, A.; Gao, C.; Song, J.B.; Wang, P.; Zhang, L.; Zhou, Y.; Shan, W.; et al. Pathogen-derived glyoxylate inhibits Tet2 DNA dioxygenase to facilitate bacterial persister formation. Cell Metab. 2025, 37, 1137–1151.e5. [Google Scholar] [CrossRef] [PubMed]
- Geyrhofer, L.; Ruelens, P.; Farr, A.D.; Pesce, D.; de Visser, J.A.G.M.; Brenner, N. Minimal Surviving Inoculum in Collective Antibiotic Resistance. mBio 2023, 14, e0245622. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I.; Lipsitch, M. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef] [PubMed]
- M100-Ed34; Performance Standards for Antimicrobial Susceptibility Testing. Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2024.
- Ferrara, A.; Grassi, G.; Grassi, F.A.; Piccioni, P.D.; Gialdroni Grassi, G. Bactericidal activity of meropenem and interactions with other antibiotics. J. Antimicrob. Chemother. 1989, 24 (Suppl. A), 239–250. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48 (Suppl. 1), 5–16, Erratum in J. Antimicrob. Chemother. 2002, 49, 1049. [Google Scholar] [CrossRef]
- Niu, H.; Gu, J.; Zhang, Y. Bacterial persisters: Molecular mechanisms and therapeutic development. Signal Transduct. Target. Ther. 2024, 9, 174. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Yoshioka, Y.; Morikawa, K.; Ariyoshi, W.; Yamasaki, R. Glucose Supplementation Enhances the Bactericidal Effect of Penicillin and Gentamicin on Streptococcus sanguinis Persisters. Antibiotics 2025, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Attrill, E.L.; Łapińska, U.; Westra, E.R.; Harding, S.V.; Pagliara, S. Slow growing bacteria survive bacteriophage in isolation. ISME Commun. 2023, 3, 95. [Google Scholar] [CrossRef] [PubMed]
- Amankwah, S.; Abdella, K.; Kassa, T. Bacterial Biofilm Destruction: A Focused Review on The Recent Use of Phage-Based Strategies with Other Antibiofilm Agents. Nanotechnol. Sci. Appl. 2021, 14, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.W.; Fung, D.K.; Wang, J.D. Regulatory Themes and Variations by the Stress-Signaling Nucleotide Alarmones (p)ppGpp in Bacteria. Annu. Rev. Genet. 2021, 55, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.W.; Proshkin, S.; Duan, W.; Epshtein, V.; Gowder, M.; Bharati, B.K.; Afanaseva, E.; Mironov, A.; Serganov, A.; Nudler, E. Control of transcription elongation and DNA repair by alarmone ppGpp. Nat. Struct. Mol. Biol. 2023, 30, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Microbiol. 2014, 12, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Coque, T.M.; de la Cruz, F. Ecology and evolution as targets: The need for novel eco-evo drugs and strategies to fight antibiotic resistance. Antimicrob. Agents Chemother. 2011, 55, 3649–3660. [Google Scholar] [CrossRef] [PubMed]
- Meylan, S.; Andrews, I.W.; Collins, J.J. Targeting antibiotic tolerance, pathogen by pathogen. Cell 2018, 172, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, X.Y.; Wan, L.G.; Jiang, W.Y.; Li, F.Q.; Yang, J.H. Efflux system overexpression and decreased OprD contribute to the carbapenem resistance among extended-spectrum beta-lactamase-producing Pseudomonas aeruginosa isolates from a Chinese university hospital. Microb. Drug Resist. 2013, 19, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, D.; Das Talukdar, A.; Choudhury, M.D.; Maurya, A.P.; Paul, D.; Chanda, D.D.; Chakravorty, A.; Bhattacharjee, A.; Chang, Y.-F. Transcriptional analysis of MexAB-OprM efflux pumps system of Pseudomonas aeruginosa and its role in carbapenem resistance in a tertiary referral hospital in India. PLoS ONE 2015, 10, e0133842. [Google Scholar] [CrossRef] [PubMed]
- Mirsalehian, A.; Kalantar-Neyestanaki, D.; Nourijelyani, K.; Asadollahi, K.; Taherikalani, M.; Emaneini, M.; Jabalameli, F. Detection of AmpC-betalactamases producing isolates among carbapenem resistant P. aeruginosa isolated from burn patient. Iran. J. Microbiol. 2014, 6, 306–310. [Google Scholar] [PubMed]
- Vestergaard, M.; Paulander, W.; Marvig, R.L.; Clasen, J.; Jochumsen, N.; Molin, S.; Jelsbak, L.; Ingmer, H.; Folkesson, A. Antibiotic combination therapy can select for broad-spectrum multidrug resistance in Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2016, 47, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Srikumar, R.; Paul, C.J.; Poole, K. Influence of mutations in the mexR repressor gene on expression of the MexA-MexB-oprM multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 2000, 182, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-resistant Pseudomonas aeruginosa: Clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.-Y.; Qu, T.-T.; Shen, P.; Wei, Z.-Q.; Yu, Y.-S.; Li, L.-J. Molecular epidemiology and mechanisms of carbapenem resistance in Pseudomonas aeruginosa isolates from Chinese hospitals. Int. J. Antimicrob. Agents 2010, 35, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Diene, S.M.; L’hOmme, T.; Bellulo, S.; Stremler, N.; Dubus, J.-C.; Mely, L.; Leroy, S.; Degand, N.; Rolain, J.-M. ISPa46, a novel insertion sequence in the oprD porin gene of an imipenem-resistant Pseudomonas aeruginosa isolate from a cystic fibrosis patient in Marseille, France. Int. J. Antimicrob. Agents 2013, 42, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Abisado-Duque, R.G.; Townsend, K.A.; Mckee, B.M.; Woods, K.; Koirala, P.; Holder, A.J.; Craddock, V.D.; Cabeen, M.; Chandler, J.R.; Mullineaux, C.W. An Amino Acid Substitution in Elongation Factor EF-G1A Alters the Antibiotic Susceptibility of Pseudomonas aeruginosa LasR-Null Mutants. J. Bacteriol. 2023, 205, e0011423. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Han, L.; Xue, Y.; Yang, I.T.; Fan, X.; Tang, R.; Zhang, C.; Zhu, M.; Tian, X.; Shao, P.; et al. Multidrug-resistant Pseudomonas aeruginosa is predisposed to lasR mutation through up-regulated activity of efflux pumps in non-cystic fibrosis bronchiectasis patients. Front. Cell Infect. Microbiol. 2022, 12, 934439. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Yang, X.; Zeng, Q.; Zhang, Y.; Li, H.; Yan, C.; Li, J.S.; Liu, H.; Du, L.; Wu, Y.; et al. Evolution of lasR mutants in polymorphic Pseudomonas aeruginosa populations facilitates chronic infection of the lung. Nat. Commun. 2023, 14, 5976. [Google Scholar] [CrossRef] [PubMed]
- Sappington, K.J.; Dandekar, A.A.; Oinuma, K.; Greenberg, E.P. Reversible signal binding by the Pseudomonas aeruginosa quorum-sensing signal receptor LasR. mBio 2011, 2, e00011-11. [Google Scholar] [CrossRef] [PubMed]
- Kok, L.-C.; Tsai, C.-C.; Liao, Y.-H.; Lo, Y.-L.; Cheng, N.-W.; Lin, C.-T.; Chang, H.-Y. Roles of transcriptional factor PsrA in the regulation of quorum sensing in Pseudomonas aeruginosa PAO1. Front. Microbiol. 2024, 15, 1424330. [Google Scholar] [CrossRef] [PubMed]
- Bondí, R.; Longo, F.; Messina, M.; D’ANgelo, F.; Visca, P.; Leoni, L.; Rampioni, G. The multi-output incoherent feedforward loop constituted by the transcriptional regulators LasR and RsaL confers robustness to a subset of quorum sensing genes in Pseudomonas aeruginosa. Mol. Biosyst. 2017, 13, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Jeske, A.; Arce-Rodriguez, A.; Thöming, J.G.; Tomasch, J.; Häussler, S. Evolution of biofilm-adapted gene expression profiles in lasR-deficient clinical Pseudomonas aeruginosa isolates. npj Biofilms Microbiomes 2022, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.H.; Hebert, W.P.; Naimie, A.; Ray, K.; Van Gelder, R.D.; DiGiandomenico, A.; Lalitha, P.; Srinivasan, M.; Acharya, N.R.; Lietman, T.; et al. Environmentally Endemic Pseudomonas aeruginosa Strains with Mutations in lasR Are Associated with Increased Disease Severity in Corneal Ulcers. mSphere 2016, 1, e00140-16. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.R.; Richardson, A.R.; Houston, L.S.; Kulasekara, H.D.; Martens-Habbena, W.; Klausen, M.; Burns, J.L.; Stahl, D.A.; Hassett, D.J.; Fang, F.C.; et al. Nutrient availability as a mechanism for selection of antibiotic tolerant Pseudomonas aeruginosa within the CF airway. PLoS Pathog. 2010, 6, e1000712. [Google Scholar] [CrossRef] [PubMed]
- Markus, V.; Golberg, K.; Teralı, K.; Ozer, N.; Kramarsky-Winter, E.; Marks, R.S.; Kushmaro, A. Assessing the Molecular Targets and Mode of Action of Furanone C-30 on Pseudomonas aeruginosa Quorum Sensing. Molecules 2021, 26, 1620. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zou, H.; Li, J.; Song, T.; Lv, W.; Wang, W.; Wang, Z.; Tao, S. Impact of quorum sensing signaling molecules in gram-negative bacteria on host cells: Current understanding and future perspectives. Gut Microbes 2022, 14, 2039048. [Google Scholar] [CrossRef] [PubMed]
- Tamber, S.; Ochs, M.M.; Hancock, R.E. Role of the novel OprD family of porins in nutrient uptake in Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Gyger, J.; Torrens, G.; Cava, F.; Bernhardt, T.G.; Fumeaux, C. A potential space-making role in cell wall biogenesis for SltB1 and DacB revealed by a beta-lactamase induction phenotype in Pseudomonas aeruginosa. mBio 2024, 15, e0141924. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, J.F.; Lamers, R.P.; Scheurwater, E.M.; Matos, A.L.; Burrows, L.L. Changes to its peptidoglycan-remodeling enzyme repertoire modulate β-lactam resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 3078–3084. [Google Scholar] [CrossRef] [PubMed]
- Lamers, R.P.; Nguyen, U.T.; Nguyen, Y.; Buensuceso, R.N.; Burrows, L.L. Loss of membrane-bound lytic transglycosylases increases outer membrane permeability and β-lactam sensitivity in Pseudomonas aeruginosa. Microbiologyopen 2015, 4, 879–895. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, B.; Fauvart, M.; Michiels, J. Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol. Rev. 2017, 41, 219–251. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Zhao, Z.; Li, Y.; Zou, J.; Ma, Q.; Zhao, Y.; Ke, Y.; Zhu, Y.; Chen, H.; Baker, M.A.; et al. Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol. Cell. 2016, 62, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 2011, 473, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Dörr, T.; Vulić, M.; Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010, 8, e1000317. [Google Scholar] [CrossRef] [PubMed]
- Vega, N.M.; Allison, K.R.; Khalil, A.S.; Collins, J.J. Signaling-mediated bacterial persister formation. Nat. Chem. Biol. 2012, 8, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Molla, M.N.; Cantor, C.R.; Collins, J.J. Bacterial charity work leads to population-wide resistance. Nature 2010, 467, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Hernando-Amado, S.; Laborda, P.; Valverde, J.R.; Martínez, J.L. Mutational background influences P. aeruginosa ciprofloxacin resistance evolution but preserves collateral sensitivity robustness. Proc. Natl. Acad. Sci. USA 2024, 121, e2109370119. [Google Scholar] [CrossRef]
- Ikawa, Y.; Wakai, T.; Funahashi, H.; Soe, T.H.; Watanabe, K.; Ohtsuki, T. Evolution of the Pseudomonas aeruginosa mutational resistome in an international Cystic Fibrosis clone. Sci. Rep. 2023, 13, 13123. [Google Scholar] [CrossRef]
- Ye, C.; Wang, A.; Breakwell, C.; Tan, R.; Bezzu, C.G.; Hunter-Sellars, E.; Williams, D.R.; Brandon, N.P.; Klusener, P.A.A.; Kucernak, A.R.; et al. Antibiotic collateral sensitivity is contingent on the repeatability of evolution. Nat. Commun. 2022, 13, 3340. [Google Scholar] [CrossRef]
- Choi, K.H.; Schweizer, H.P. mini-Tn7 insertion in bacteria with single attTn7 sites: Example Pseudomonas aeruginosa. Nat. Protoc. 2006, 1, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Heurlier, K.; Williams, F.; Heeb, S.; Dormond, C.; Pessi, G.; Singer, D.; Cámara, M.; Williams, P.; Haas, D. Positive control of swarming, rhamnolipid synthesis, and lipase production by the posttranscriptional RsmA/RsmZ system in Pseudomonas aeruginosa PAO1. J. Bacteriol. 2004, 186, 2936–2945. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).