1. Introduction
Respiratory syncytial virus (RSV) is a leading cause of severe lower respiratory tract infections in infants, the elderly, and immunocompromised individuals [
1]. Despite decades of research, effective antiviral therapies remain limited, highlighting the urgent need to elucidate key mechanisms underlying RSV pathogenesis. Emerging evidence points to virus–host cytoskeletal interactions as critical drivers of RSV assembly, morphogenesis, and intercellular spread [
2].
RSV actively manipulates host cytoskeletal components—namely actin filaments, microtubules, and intermediate filaments—to support filamentous virion formation and efficient viral transmission [
3,
4,
5]. The filamentous morphology of RSV particles has been associated with increased infectivity and enhanced syncytium formation, facilitating direct cell-to-cell dissemination [
6]. Notably, RSV activates the ARP2/3 complex to promote actin polymerization and filopodia formation in infected A549 cells, enhancing viral budding and spread [
7]. RSV also disrupts F-actin structures and reduces the actin-binding protein cortactin, compromising epithelial barrier function [
8].
Key regulators of cytoskeletal remodeling include the Rho family of small GTPases—RhoA, Rac1, and Cdc42—which orchestrate actin dynamics, cell morphology, and membrane organization [
9]. RhoA, in particular, is activated in RSV-infected cells and localizes within lipid rafts—cholesterol- and sphingolipid-rich membrane microdomains that facilitate signal transduction and cytoskeletal regulation [
10]. The disruption of lipid rafts perturbs RhoA activation and downstream cytoskeletal functions. Additionally, RSV exploits Rab11-dependent microtubule and actin-mediated trafficking via Rab11 family interacting proteins (Rab11-FIPs), enhancing intracellular viral transport [
11].
Previous studies using C3 toxin—a broad-spectrum Rho inhibitor—highlighted the role of RhoA in filamentous virion formation and syncytium induction [
10]. However, the C3 toxin’s clinical utility is hampered by nonspecific cytotoxicity, poor cell permeability, and pan-Rho inhibition that complicates data interpretation and reduces clinical applicability. In contrast, our study used Rhosin, a small-molecule RhoA-specific inhibitor developed through in silico virtual screening and structure-based design, specifically targeting the GEF–RhoA interaction site [
12]. It exhibits enhanced selectivity, cellular uptake, and reduced cytotoxicity, making it a promising tool for dissecting RhoA-specific pathways and improving therapeutic potential, making it a more suitable candidate for potential clinical applications [
12]. Consistent with this, our MTT-based cytotoxicity assay (
Figure 1) confirmed that Rhosin maintains high cell viability compared to C3 toxin, validating its use for exploring host–virus interactions and as a potential therapeutic approach.
Building on Gower et al.’s findings, we employed Rhosin to selectively inhibit RhoA and investigate its role in RSV-induced morphological and functional changes. Our data demonstrate that RhoA inhibition does not significantly impair viral replication (as determined by PFU counts) but markedly alters virion morphology and cell fusion dynamics. Scanning electron microscopy revealed that Rhosin-treated cells exhibited a marked reduction in filamentous virions compared to the large aggregates of long filaments typically observed in untreated cells. Complementary analyses using sucrose gradient velocity sedimentation, fluorescence microscopy, and transmission electron microscopy further confirmed that RhoA inhibition shifts the balance from filamentous to spherical RSV particles. Immunofluorescence studies using the raft-selective dye DiIC16(3) and anti-F protein antibodies revealed that Rhosin disrupts the association of RSV fusion proteins with lipid rafts, implicating RhoA in raft-dependent virion assembly.
Together, these findings implicate RhoA as a critical host factor for filamentous virion formation, syncytium development, and efficient RSV dissemination. Targeting host signaling pathways that modulate viral morphology—rather than replication alone—offers a novel therapeutic avenue to attenuate RSV spread. This study contributes to our understanding of virus–host interactions and identifies RhoA as a promising target for anti-RSV strategies.
2. Materials and Methods
2.1. Virus Propagation and Cell Culture
The A2 strain of RSV was obtained from the American Type Culture Collection, Manassas, VA, USA (ATCC; VR-1540). RSV A2 stocks were propagated in HEp-2 cells, and viral titers were determined by plaque-forming unit (PFU) assays [
13] to ensure consistent infection doses across experiments.
HEp-2 cells (ATCC; CCL-23) and HEK-293T cells (ATCC; CRL-3216) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37 °C in a humidified 5% CO2 atmosphere.
2.2. Comparative Cytotoxicity of Rhosin and C3 Toxin
Cell viability in response to Rhosin treatment (20 µM) was assessed using trypan blue exclusion and MTT assays at 12, 24, and 48 h post-treatment. No significant cytotoxicity was observed at this concentration.
Given the known limitations of the C3 toxin in terms of cytotoxicity and cell permeability, we undertook a comparative analysis of Rhosin, a specific RhoA inhibitor, and C3 toxin cytotoxicity in HEp-2 cells to establish a safer and more specific approach for RhoA inhibition in RSV research. Briefly, HEp-2 cells were seeded at 1 × 104 cells per well in 96-well plates and were allowed to adhere overnight. To evaluate cytotoxicity, the cells were treated with increasing concentrations of Rhosin (0, 5, 10, 20, and 40 µM; Sigma-Aldrich, Saint Louis, MO, USA) or C3 toxin (0, 1, 2.5, 5, and 10 µg/mL; Cytoskeleton Inc., Denver, CO, USA) for 24, 48, and 72 h. DMSO and PBS served as vehicle controls for Rhosin and the C3 toxin, respectively. Following treatment, cell viability was assessed using an MTT assay by adding MTT reagent (0.5 mg/mL) for 3 h at 37 °C. After incubation, the media were removed, and the resulting formazan crystals were solubilized in DMSO. Absorbance was measured at 570 nm using a microplate reader, and cell viability was expressed as a percentage relative to untreated controls. All treatments were performed in triplicate, and the data were presented as the mean ± standard deviation.
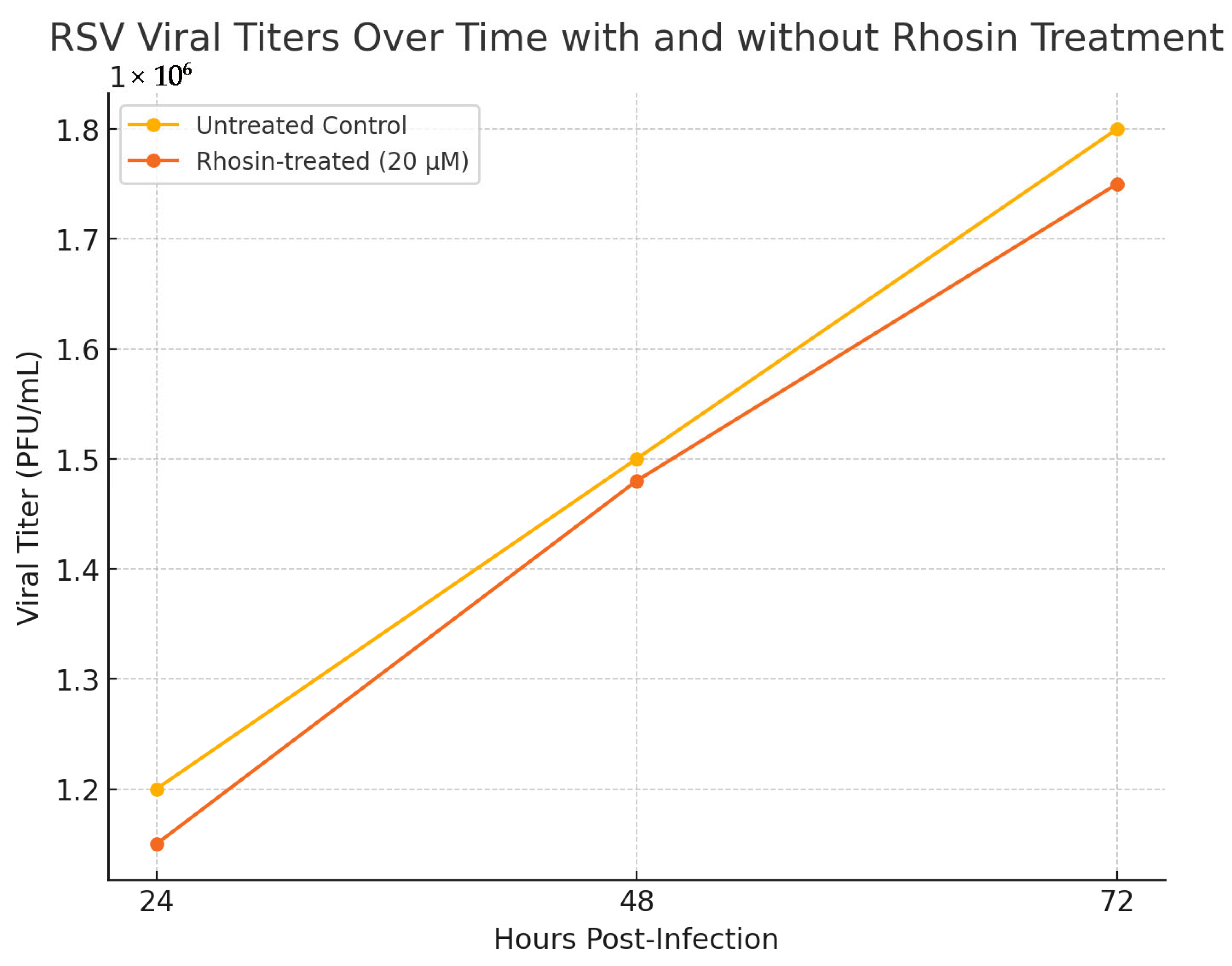
2.3. RSV Replication Kinetics in Rhosin-Treated Cells
To assess the reproducibility and temporal impact of Rhosin treatment on RSV replication, HEp-2 cells were infected with RSV at a multiplicity of infection (MOI) of 1 and treated with 20 µM Rhosin. Supernatants were collected at 24, 48, and 72 h post-infection, and viral titers were determined using plaque-forming unit (PFU) assays as previously described ([
13,
14], and
Section 2.4). Untreated infected cells served as controls. Each condition was performed in triplicate, and the data were analyzed using two-way ANOVA to compare viral titers across treatment groups and time points.
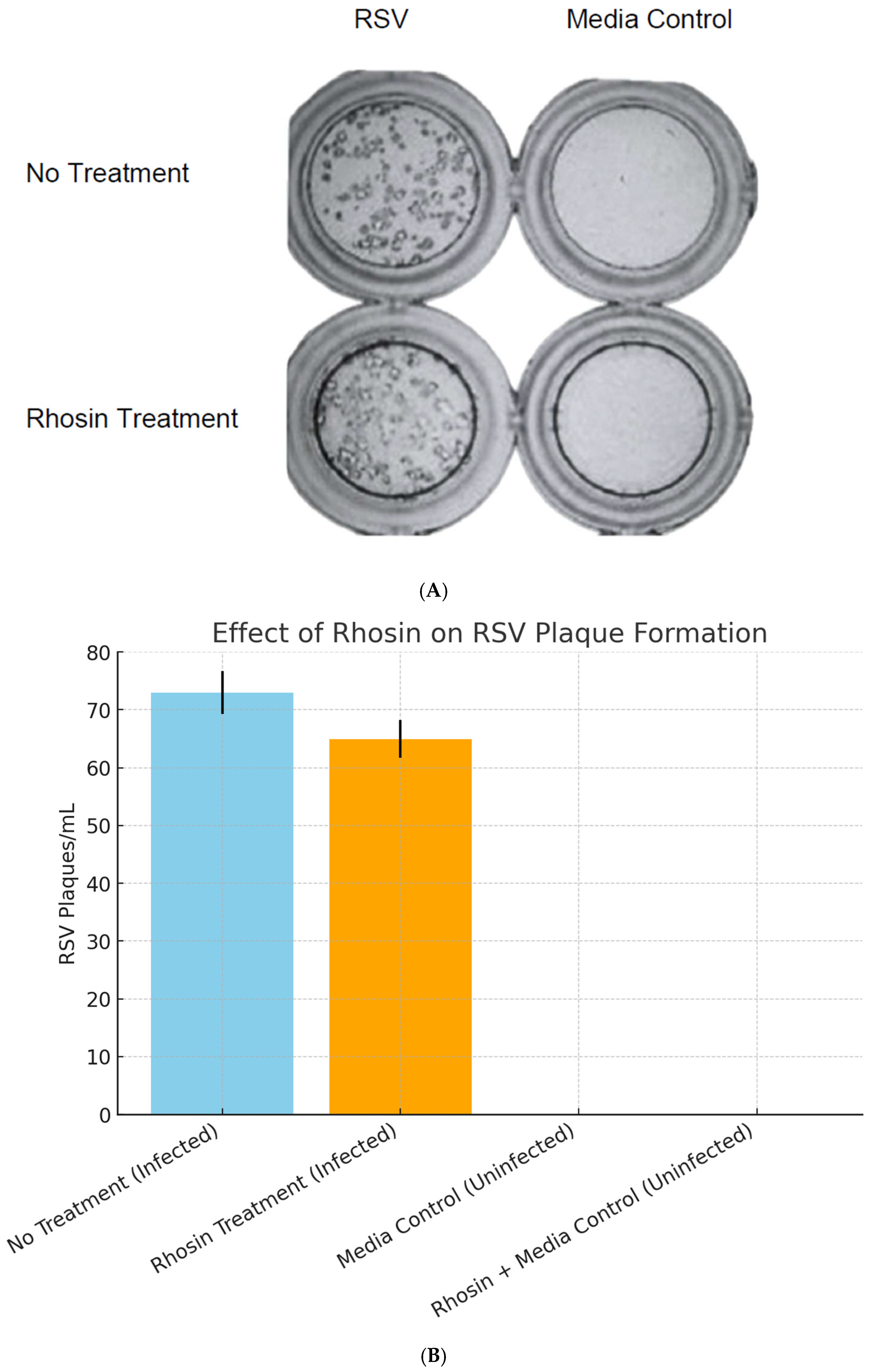
2.4. Effect of Rhosin Treatment on RSV Replication and Plaque Formation in HEp-2 Cells
For infection studies, HEp-2 cells were seeded at appropriate densities and infected with RSV at a MOI of 1 for 1 h. To investigate the role of RhoA, the cells were treated with Rhosin at 20 µM concentrations determined by preliminary dose–response studies. Rhosin was added to the culture medium prior to infection and maintained throughout the infection period (72 h). Control groups were treated with vehicle alone. Viral replication was assessed using a standard PFU assay [
13]. Briefly, after a defined incubation period post-infection, the supernatants were collected and serially diluted. Diluted samples were overlaid onto approximately 1 × 10
5 HEp-2 cell monolayers and incubated for 1 h before the agarose overlay was applied. After 72 h, plates were fixed with methanol, and RSV-specific immunoperoxidase staining was performed [
14]. RSV plaques were counted in each well to quantify infectious virions, comparing Rhosin-treated samples with untreated controls.
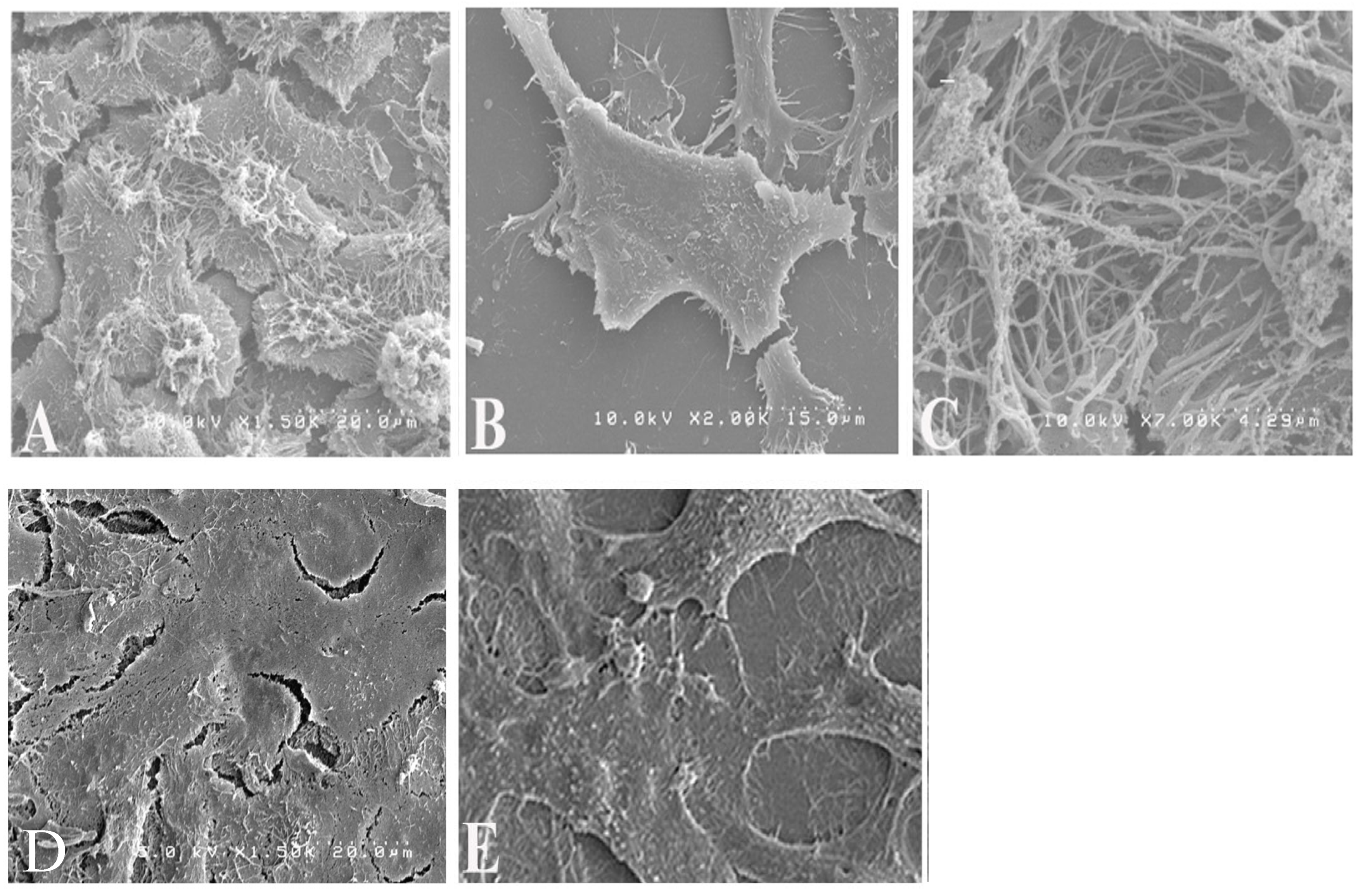
2.5. Scanning Electron Microscopy (SEM)
To examine virion morphology, HEp-2 cells on 12 mm coverslips were pre-treated for 16 h with 20 µM Rhosin or mock-treated and then infected with RSV (MOI 1), with treatments maintained post-adsorption. Controls included uninfected untreated cells. At 48 h post-infection, the cells were fixed in 4% glutaraldehyde (1 h), post-fixed with 1% osmium tetroxide (15 min), dehydrated through graded ethanol washes, critical point dried, sputter coated with gold, and imaged using Hitachi S4200 SEM, Pleasanton, CA, USA. SEM images were captured to visualize the differences in filamentous and spherical virion forms between Rhosin-treated and untreated cells.
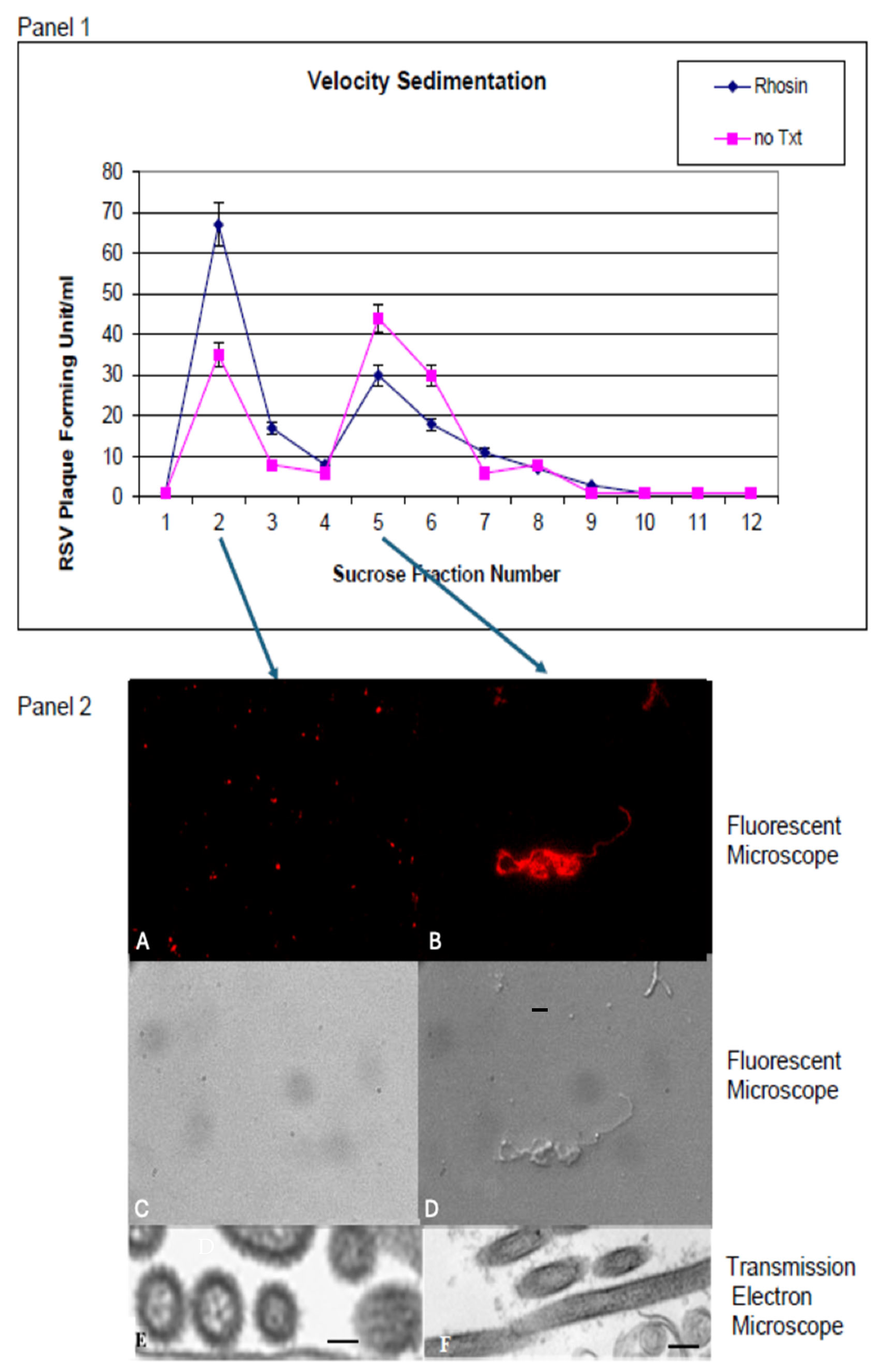
2.6. Sucrose Gradient Velocity Sedimentation
Sucrose gradient sedimentation was employed to separate RSV particles based on density and shape. HEp-2 cells in 25-cm
2 flasks were either left untreated or treated with 20 µM Rhosin for 24 h and then infected with RSV (MOI of 0.5). After a 72 h infection, the infected cells were harvested and layered over a continuous 15 to 60% sucrose gradient. The virus was then separated by velocity centrifugation at 14,000 rpm for 10 min at 4 °C in a Sorvall Sure Spin 630 centrifuge, Sorvall Thermo Scientific, Waltham, MA, USA. Two-milliliter fractions were collected, and plaque assays were performed with the fractions as described previously [
13]. Fractions were also analyzed by fluorescence microscopy and transmission electron microscopy (TEM) to distinguish between spherical and filamentous virions, thereby quantifying the impact of RhoA inhibition on particle morphology.
2.7. Fluorescence Microscopy and Transmission Electron Microscopy (TEM)
Fluorescence microscopy was used to assess the distribution and localization of RSV proteins in infected cells. Specimens for fluorescence microscopy were prepared on coverslips and stained. Briefly, 50 µL of RSV-containing supernatant from infected cells was smeared on a coverslip and allowed to air dry. The virus particles were fixed with 3.7% formaldehyde in PBS for 30 min. The samples were washed twice with PBS-Tween 20 and blocked with 5% nonfat dry milk in PBS for 30 min. The cells were stained with a 1:500 dilution of anti-RSV antibody (Maine Biotechnology Services, Portland, ME, USA) prepared in 1% nonfat dry milk and incubated for 1 h at room temperature. This was followed by staining with a 1:1000 dilution of goat anti-mouse IgG (H + L) secondary antibody conjugated to Rhodamine (ThermoFisher Scientific, Waltham, MA, USA), also for 1 h at room temperature. After three washes with PBS-Tween 20, the coverslips were mounted on microscope slides. The specimens were viewed with a Zeiss AxioPlan 2 fluorescence microscope, Carl Zeiss Microscopy, Dublin, CA, USA and pictures were taken with a Hamamatsu ORCA-ER digital camera, Hamamatsu Corporation, Bridgewater, NJ, USA.
Additionally, TEM provided high-resolution images of virion ultrastructure. For evaluations of particles separated by velocity sedimentation, the bands associated with the peak fractions were concentrated by centrifugation onto a 60% sucrose cushion. A sample from the concentrated band was applied to a grid, stained with 0.5% uranyl acetate, and examined using a Hitachi H-7000 transmission electron microscope, Hitachi High-TechAmerica, Inc., Schaumburg, IL, USA, allowing for detailed visualization of RSV morphology in both Rhosin-treated and untreated conditions.
2.8. Beta-Galactosidase Cell-to-Cell Fusion Assay
The functional impact of altered virion morphology on cell-to-cell fusion was measured using a beta-galactosidase reporter assay [
15]. Briefly, HEp-2 cells were divided into two populations:
A. Effector cells were infected with vaccinia virus vTF7-3 (MOI = 10) to express T7 polymerase, and then transfected with plasmids for RSV glycoproteins F, G, SH, and M (under T7 promoter control). At 5 h post-transfection, the cells were trypsinized, resuspended at 2 × 10
7 cells/mL in MEM containing 2.5% FBS, and 20 μg/mL of either the Rhosin or the control media, or left untreated, incubated overnight at 32 °C, and later adjusted to 1 × 10
6 cells/mL in Opti-MEM.
B. Reporter cells were infected with a recombinant vaccinia virus engineered to express β-galactosidase under the T7 promoter (provided by E.A. Berger, National Institutes of Health, Bethesda, MD, USA). At 5 h post-infection, these cells were trypsinized and suspended in Opti-MEM at a concentration of 1 × 10
6 cells/mL.
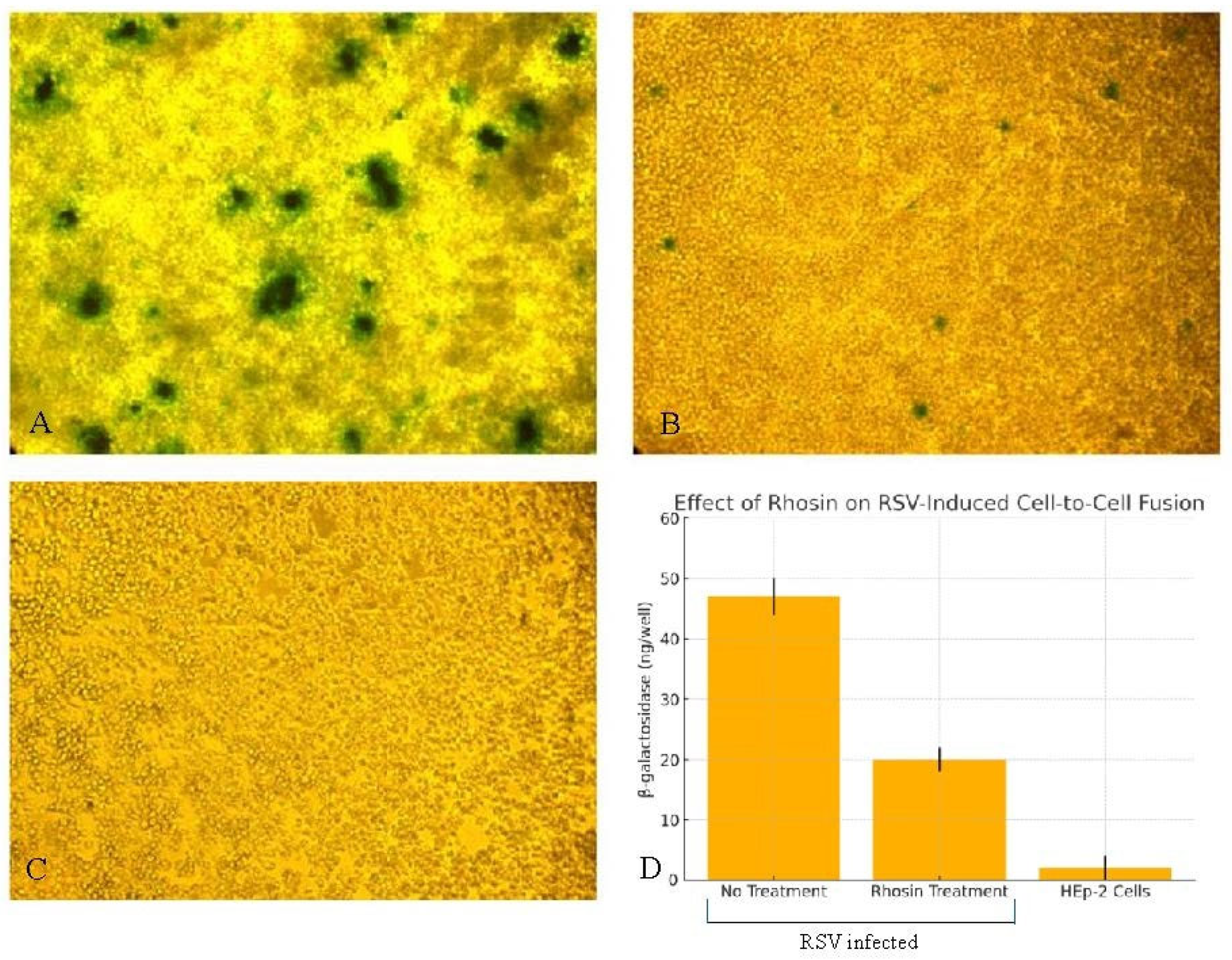
Equal volumes (100 μL) of the effector and reporter cell suspensions were mixed in triplicate wells in a 96-well tissue culture plate and incubated at 37 °C for 4 h to allow for cell-to-cell fusion. Two complementary approaches were used to measure fusion events: 1. Colorimetric Lysate Assay: β-galactosidase activity was quantified using the CPRG (chlorophenol red-β-D-galactopyranoside) assay kit (G-Biosciences, Saint Louis, MO, USA, Cat. #786-651), which provides a sensitive colorimetric readout at 570–595 nm. Infected or transfected HEp-2 cells were lysed with Mammalian Cell PE LB™ buffer (G-Biosciences, Saint Louis, MO, USA, Cat. #786-180), freeze-thawed, and clarified by centrifugation. In total, 20 µL of the clarified lysate was added to a 96-well plate and incubated with 130 µL of 1× CPRG substrate at 37 °C. Reactions were stopped after color development using the provided stop solution, and absorbance was read in triplicate using a microplate reader. The readings were normalized to mock-transfected controls and the protein concentration of lysates. β-galactosidase activity was calculated based on a chlorophenol red standard curve and expressed as ng of product per well. 2. In Situ X-Gal Staining: Fixed cells were stained with an X-Gal solution, and blue syncytia were visualized under phase-contrast microscopy. This dual-assay approach allowed for both quantitative measurement and visual confirmation of RSV F-induced cell-to-cell fusion and facilitated the assessment of the inhibitory effects of the Rhosin on fusion events.
2.9. Synthetic RSV Protein Expression and Transfection
Briefly, protein sequences corresponding to the RSV A2 strain structural proteins fusion (F), attachment glycoprotein (G), small hydrophobic protein (SH), and matrix (M), as documented in GenBank: KT992094.1 [
16], were reverse-translated into human cell-preferred codons using the Wisconsin Genetics Computer Group (GCG) software package version 10.3. Subsequently, oligonucleotides covering these four genes were synthesized commercially by Sigma-Genosys, with each oligonucleotide being 75 bases long with an overlap of 25 bases between consecutive segments. The codon-optimized genes were then assembled into expression vectors derived from the pNGVL-3 plasmid backbone and verified through sequencing to confirm accuracy and integrity. Plasmid DNA was purified by double cesium chloride gradient ultracentrifugation to ensure high purity suitable for transfection. The cells were transfected with 2 µg each of the plasmids encoding the F, G, SH, and M proteins (totaling 8 µg DNA per transfection) using the calcium phosphate precipitation method described by Chen and Okayama [
17].
2.10. Immunofluorescence and Lipid Raft Labeling
To investigate the association between RSV structural proteins and lipid raft microdomains, we employed immunofluorescence microscopy coupled with selective lipid raft labeling. DiIC
16(3) (1,1-dihexadecyl-3,3,3,3-tetramethylindocarbocyanine perchlorate) consists of a long acyl chain, which allows for its incorporation into the more rigid, ordered lipid domains, compared to DiIC
12(3) (1,1-didodecyl-3,3,3,3-tetramethylindocarbocyanine perchlorate), which labels the fluid non-raft membrane components [
18]. Briefly, HEK-293T cells were seeded on coverslips and transfected with 2 µg each of the plasmids encoding the F, G, SH, and M proteins (totaling 8 µg DNA per transfection) using the calcium phosphate precipitation method described by Chen and Okayama [
17]. The vector backbone without viral inserts was used as filler DNA to maintain consistent total DNA amounts across experimental conditions. At 48 h post-transfection, cells seeded on coverslips were chilled on ice and incubated with a 1:100 dilution of lipid raft-selective dye DiIC
16(3) or non-raft-selective dye DiIC
12(3) dissolved in ethanol for 15 min. Subsequently, the cells were fixed with 3.7% formaldehyde for 10 min at room temperature, permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 10 min and blocked with 5% nonfat milk in PBS for 15 min to minimize nonspecific binding. The cells were then incubated for 1 h at room temperature with primary antibodies: mouse anti-RSV F monoclonal antibody (Millipore Sigma, Burlington, MA, USA), diluted at 1:500 in 3% milk-PBS. Following washing with PBS-Tween-20, the cells were incubated for 1 h with Alexa Fluor 488-conjugated secondary antibodies (anti-mouse IgG, Molecular Probes, Eugene, OR, USA) diluted to 1:1000 in 3% milk–PBS. Images were captured using a Zeiss Axioplan fluorescence microscope, Carl Zeiss Microscopy, Dublin, CA, USA, equipped with a 100× oil immersion objective, and digital images were acquired using an Axiocam camera, Carl Zeiss Microscopy, Dublin, CA, USA.
2.11. Rhosin-Mediated Disruption of RSV Protein Assembly in Lipid Rafts
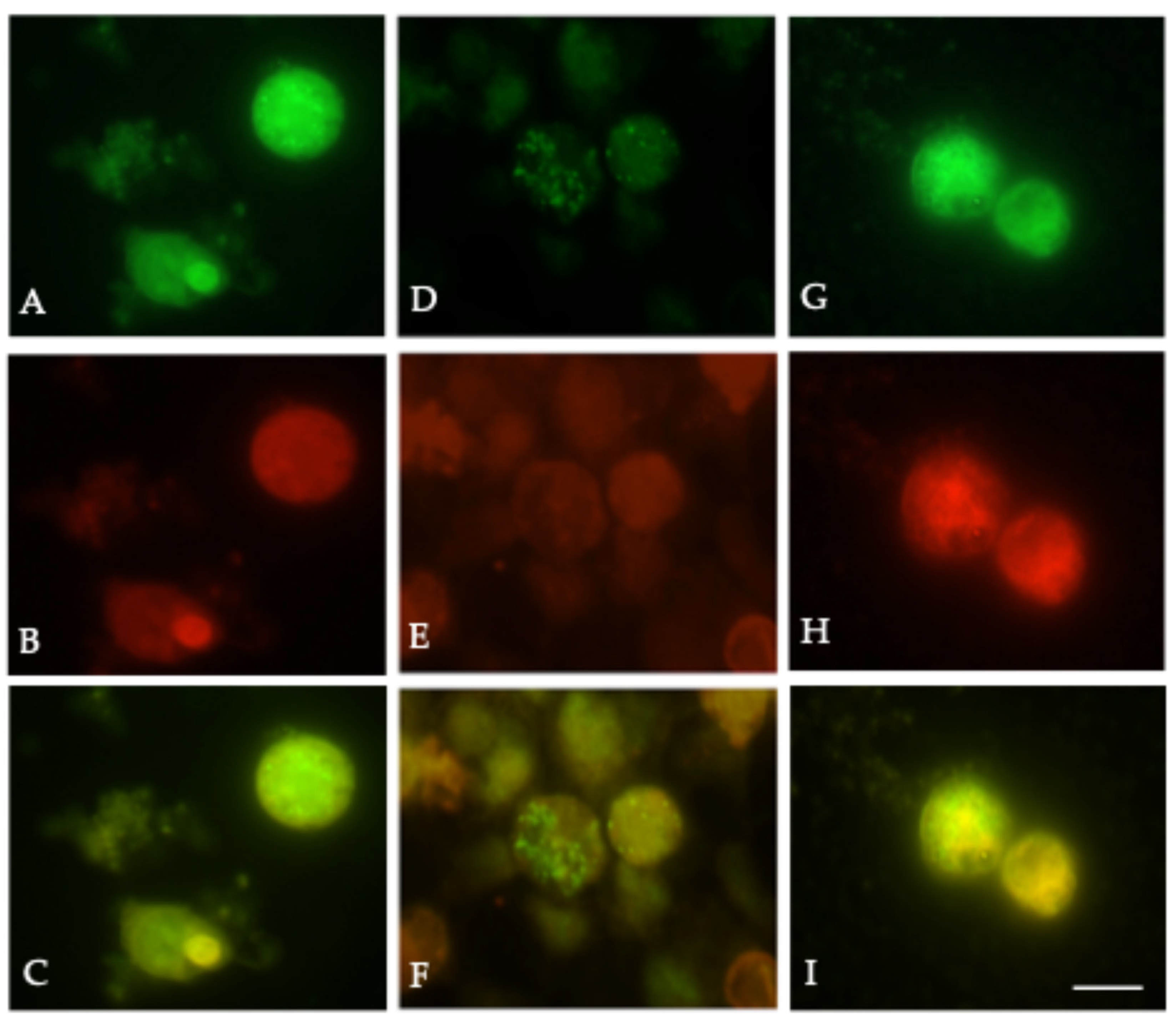
To determine the effect of RhoA inhibition in RSV structural protein assembly in lipid rafts, we used Rhosin to inhibit RhoA in HEK-293T cells. HEK-293T cells were seeded on coverslips and were either left untreated or treated with 20 µM Rhosin for 24 h and then transfected with pNGVL-3 plasmids encoding the F, G, SH, and M proteins. Treatment with 20 µM Rhosin was continued post transfection until 48 h. At 48 h post-transfection, the cells were washed twice with PBS and then labeled with dyes and antibodies as described above. The samples were mounted and imaged by fluorescence microscopy. Rhosin treatment resulted in the disruption of F protein colocalization with DiIC16 (3), supporting the role of cholesterol-rich microdomains in viral particle assembly.
2.12. Data Analysis
All experiments were conducted in triplicate. The data were analyzed using appropriate statistical methods, including t-tests or ANOVA, to compare differences between Rhosin-treated and control groups. Significance was set at p < 0.05. Data presentation included the means ± standard error of the mean (SEM), with graphical representation generated using GraphPad Prism, Version 10.5.0 or standard software packages.
Overall, these methods enabled a comprehensive evaluation of the role of RhoA in RSV infection, specifically addressing its impact on virion morphology, syncytium formation, and the efficiency of viral spread.
4. Discussion
This study highlights the critical role of RhoA signaling in regulating respiratory syncytial virus morphology, syncytium formation, and cell-to-cell fusion, and further establishes lipid rafts as critical platforms for RSV assembly and release. While RSV exists in both filamentous and spherical forms, filamentous particles are considered more effective at mediating direct intercellular spread [
10,
19]. MTT viability assays confirmed that Rhosin exhibits significantly lower cytotoxicity than C3 toxin (
Figure 1,
Table 1). Rhosin displayed minimal cytotoxic effects, maintaining high cell viability across the tested concentrations, while the C3 toxin showed pronounced, dose-dependent toxicity. These findings underscore Rhosin’s improved safety profile and specificity, supporting its application as a more suitable RhoA inhibitor for investigating RSV–host cytoskeletal interactions, and as a promising candidate for therapeutic intervention.
To further assess the reproducibility and temporal impact of Rhosin treatment on RSV replication, viral titers were measured at 24, 48, and 72 h post-infection in HEp-2 cells treated with 20 µM Rhosin or left untreated. Consistent with the viral titer analysis across multiple time points, our findings confirm that Rhosin-mediated RhoA inhibition does not impact RSV replication kinetics, underscoring that its primary effects are restricted to altering virion morphology and reducing cell-to-cell spread.
While RhoA inhibition with Rhosin did not significantly impact RSV replication, it profoundly altered virion structure by shifting RSV particles from filamentous to spherical forms, as corroborated by scanning electron microscopy (SEM), sucrose gradient velocity sedimentation, fluorescence microscopy, and transmission electron microscopy (TEM), which collectively confirmed that RhoA inhibition disrupts the formation of filamentous RSV particles (
Figure 3,
Figure 4 and
Figure 5). These findings suggest that RhoA signaling plays a pivotal role in RSV assembly and dissemination. Our data corroborates previously published results showing that blocking RhoA signaling with C3 toxin did not affect the RSV replication but affected filament formation [
10]. However, the C3 toxin has other toxic effects on host cells, and C3 is not ideal for clinical use due to its low cellular penetration. These results reinforce that RhoA signaling is indispensable for filamentous RSV assembly, a phenotype associated with enhanced cell-to-cell fusion and dissemination.
Filamentous RSV particles are thought to facilitate viral spread through enhanced cell-to-cell fusion and syncytium formation, thereby bypassing the need for extracellular release and re-entry [
19]. Filamentous morphology may allow for continuous virion transmission through intercellular junctions, bypassing immune surveillance and enhancing syncytium-mediated spread, which has been linked to increased disease severity in vivo [
19]. The functional consequences of disrupted filament formation were demonstrated using a β-galactosidase fusion assay, which showed a significant reduction in syncytium formation in Rhosin-treated cells (
Figure 6), correlating with the loss of filamentous virions observed by SEM (
Figure 3). This supports the notion that filamentous RSV is more efficient in mediating direct cell-to-cell transmission, a key factor in viral pathogenesis. The disruption of filament formation may limit RSV’s ability to spread efficiently within the host, potentially offering a novel antiviral strategy.
Despite these morphological and functional changes, plaque assays confirmed that RSV replication remained unaffected by RhoA inhibition between Rhosin-treated and control groups (
Figure 5A,B). This suggests that RhoA signaling is not essential for viral genome replication or particle production but is instead crucial for virion assembly and dissemination. Sucrose gradient velocity sedimentation further validated this, showing a distinct shift toward spherical RSV particles upon RhoA inhibition by Rhosin treatment (
Figure 4).
Our lipid raft localization studies demonstrated that the RSV F protein preferentially associates with raft-selective domains, and this association is disrupted by RhoA inhibition (
Figure 7), underscoring the requirement for intact lipid rafts in assembly. Lipid rafts are dynamic, cholesterol- and sphingolipid-enriched microdomains of the cell membranes that act as platforms for signal transduction, protein sorting, and pathogen entry and facilitate membrane curvature and scission; their disruption impairs filamentous virion formation and viral dissemination. These results are consistent with the literature showing that many enveloped viruses, including HIV, influenza, coronavirus, Ebola, and measles, hijack lipid rafts for their assembly and budding [
20,
21,
22,
23,
24,
25]. These domains concentrate viral proteins, facilitate glycoprotein clustering, and interact with host factors that coordinate membrane curvature and scission. As such, lipid rafts have emerged as strategic targets for antiviral drug development [
26,
27].
Our findings align with previous studies indicating that host cytoskeletal dynamics play a crucial role in RSV infection [
2]. The dependence of filamentous RSV on RhoA activity highlights a potential vulnerability that could be exploited therapeutically. RhoA exerts its effects through several downstream effectors, including Rhokinase/ROCK/ROK and mDia1, which are known to regulate actin cytoskeleton organization and stress fiber formation [
28,
29]. These pathways likely contribute to the structural support necessary for filamentous RSV morphogenesis. Additionally, RhoA-mediated activation of cofilin and LIM kinase pathways may influence membrane curvature and fusion protein trafficking [
30]. Targeting host factors like RhoA or its downstream effectors, rather than viral components, may reduce the likelihood of resistance development while limiting viral spread. By shifting the balance from filamentous to spherical virion formation, such interventions may reduce the virus’s ability to form syncytia and spread efficiently, thereby limiting disease progression even in the absence of a direct antiviral effect on replication. This approach could be particularly valuable in controlling RSV infections in high-risk populations, such as infants and immunocompromised individuals [
1].
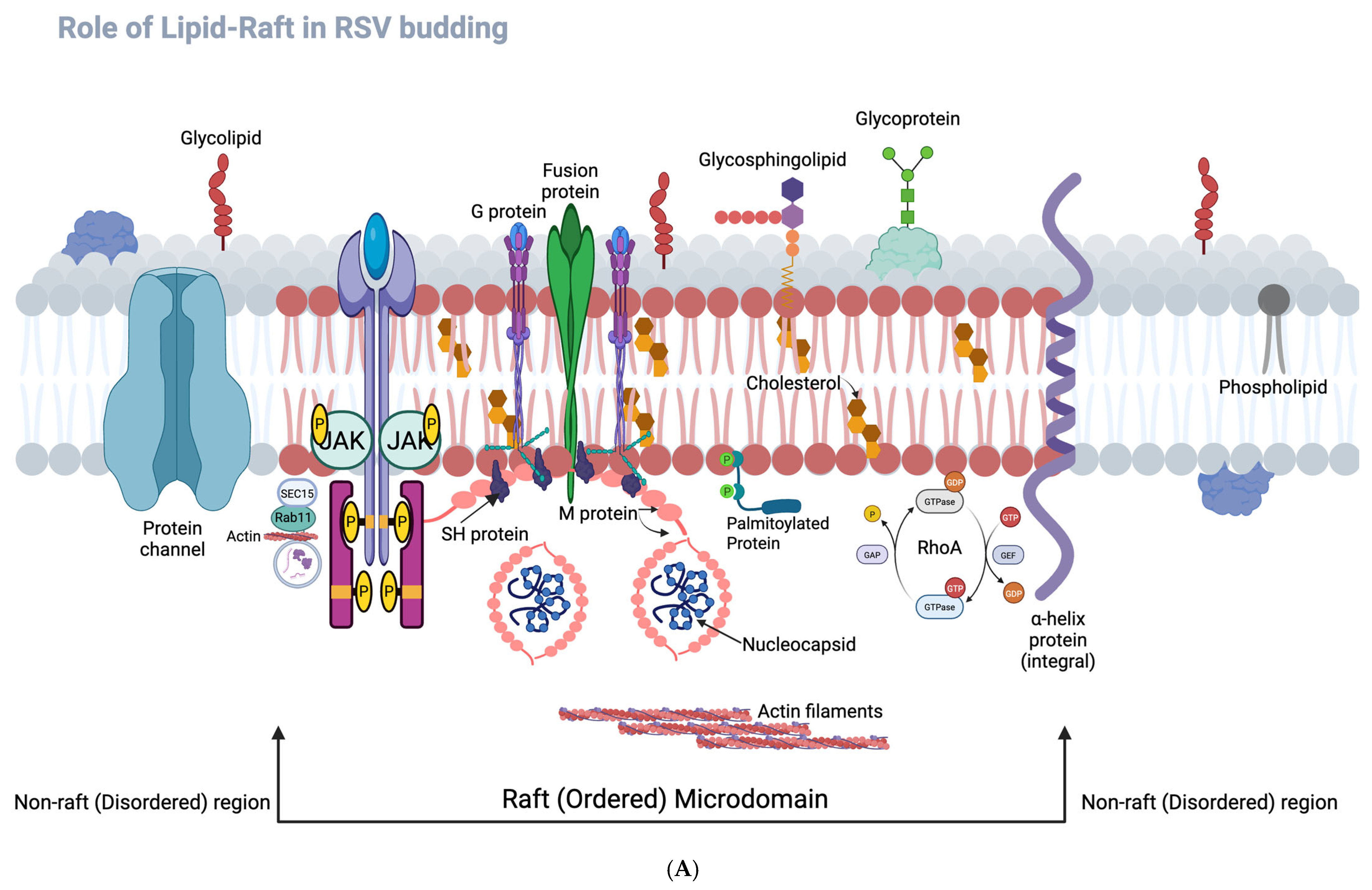
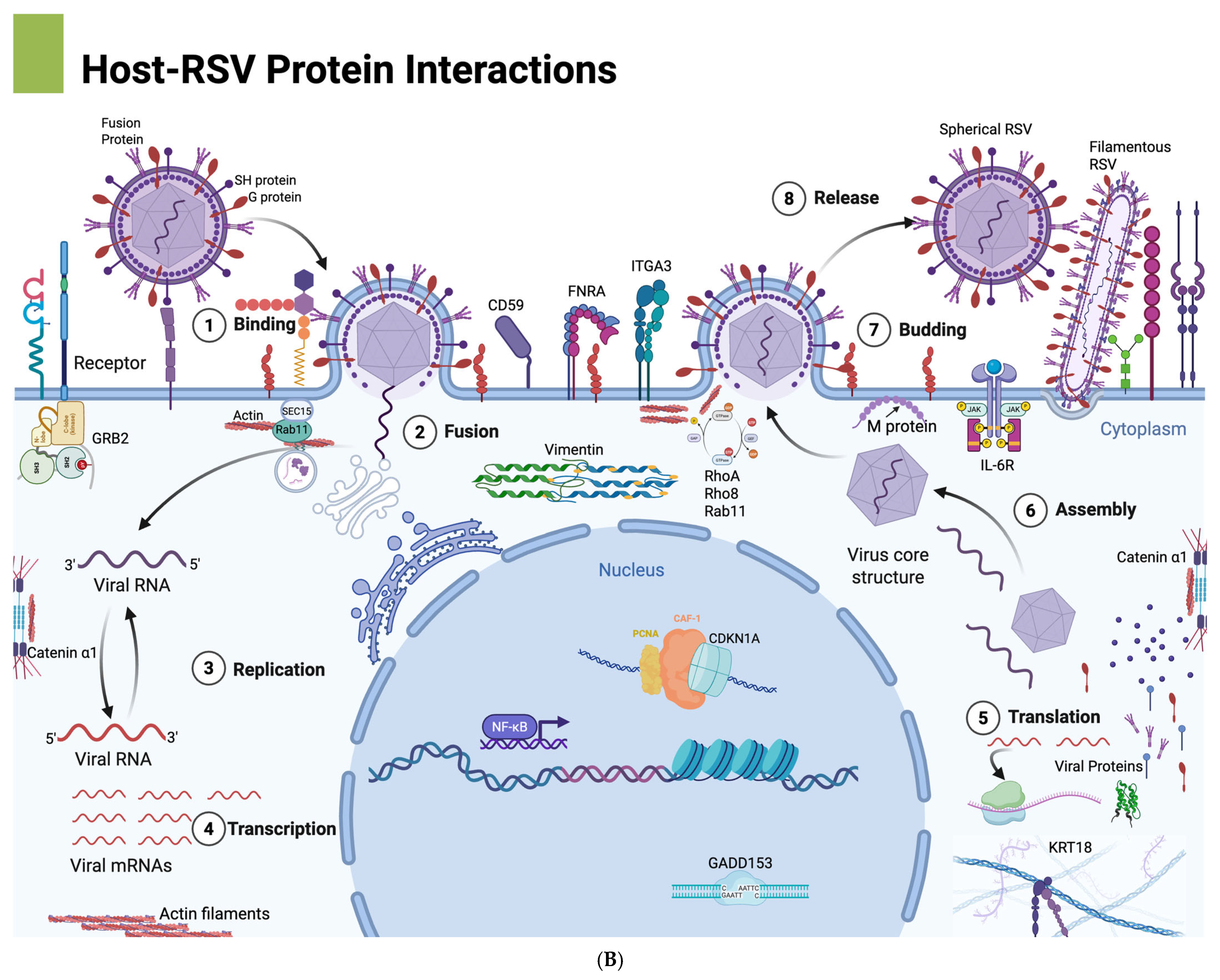
The conceptual frameworks illustrated in
Figure 8A,B further contextualize our findings.
Figure 8A highlights the spatial organization of RSV assembly at lipid raft versus non-raft membrane domains, illustrating how RSV structural proteins—particularly F, G, and M—preferentially assemble within cholesterol-rich lipid rafts to promote filamentous morphogenesis. The disruption of this organization via RhoA inhibition impairs proper membrane targeting of the F protein, likely contributing to the observed shift toward spherical virion morphology and reduced cell-to-cell fusion. Complementarily,
Figure 8B presents an overview of the RSV life cycle, integrating host cellular components that facilitate or respond to infection. It visually maps key stages including viral entry, replication, and assembly, and highlights host cytoskeletal elements (e.g., actin filaments, vimentin), inflammatory mediators (e.g., IL-6), and upregulated genes with roles in apoptosis, stress response, and membrane organization. Several of these—VIM, RHOA, FNRA, and CD59—are known to localize to lipid rafts, suggesting a coordinated host–virus interface at these membrane microdomains (our unpublished observations). Together, these schematics support a model in which RSV exploits RhoA-dependent lipid raft assembly and host cytoskeletal networks to drive filamentous virion formation and efficient spread. Although both spherical and filamentous particles utilize lipid raft components during assembly, filamentous virions exhibit a greater dependence on these microdomains.
In conclusion, our findings identify RhoA signaling as a critical regulator of RSV filamentous morphogenesis and cell-to-cell spread. RhoA coordinates the localization of RSV fusion (F) proteins to lipid raft microdomains, enabling efficient assembly of filamentous virions and promoting syncytium formation. The pharmacological inhibition of RhoA using Rhosin alters virion morphology, disrupts F protein partitioning into lipid rafts, and significantly reduces syncytium formation—all without affecting total viral replication. These results suggest that RhoA, or its downstream effectors, may represent promising targets for host-directed antiviral intervention.
However, the broader implications of RhoA inhibition on viral infectivity and transmission dynamics remain to be established, particularly in physiologically relevant in vivo models. Future studies should evaluate the impact of RhoA inhibition on RSV pathogenesis and explore potential side effects of targeting this pathway in host tissues. Additionally, identifying specific downstream effectors of RhoA involved in filament formation may yield novel antiviral strategies that selectively impair RSV assembly and spread.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}