
Genetic Diversification of Tomato and Agricultural Soil Management Shaped the Rhizospheric Microbiome of Tomato (Solanum lycopersicum)

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Phylogenetic Analysis
2.3. Experimental Design
2.4. DNA Isolation, Amplicon Sequencing, and Bioinformatic Analysis
2.5. Taxonomic, Alpha, and Beta Diversity
2.6. Determination of Specific Taxa Patterns by Genotype and Functional Prediction
2.7. Co-Occurrence Network Analysis
2.8. Statistical Analysis
3. Results
3.1. Phylogeny of the Selected Cultivars
3.2. Effect of Plant Genetic Diversity and Soil Type on the Tomato Rhizosphere-Associated Microbiota
3.2.1. Taxonomical Diversity
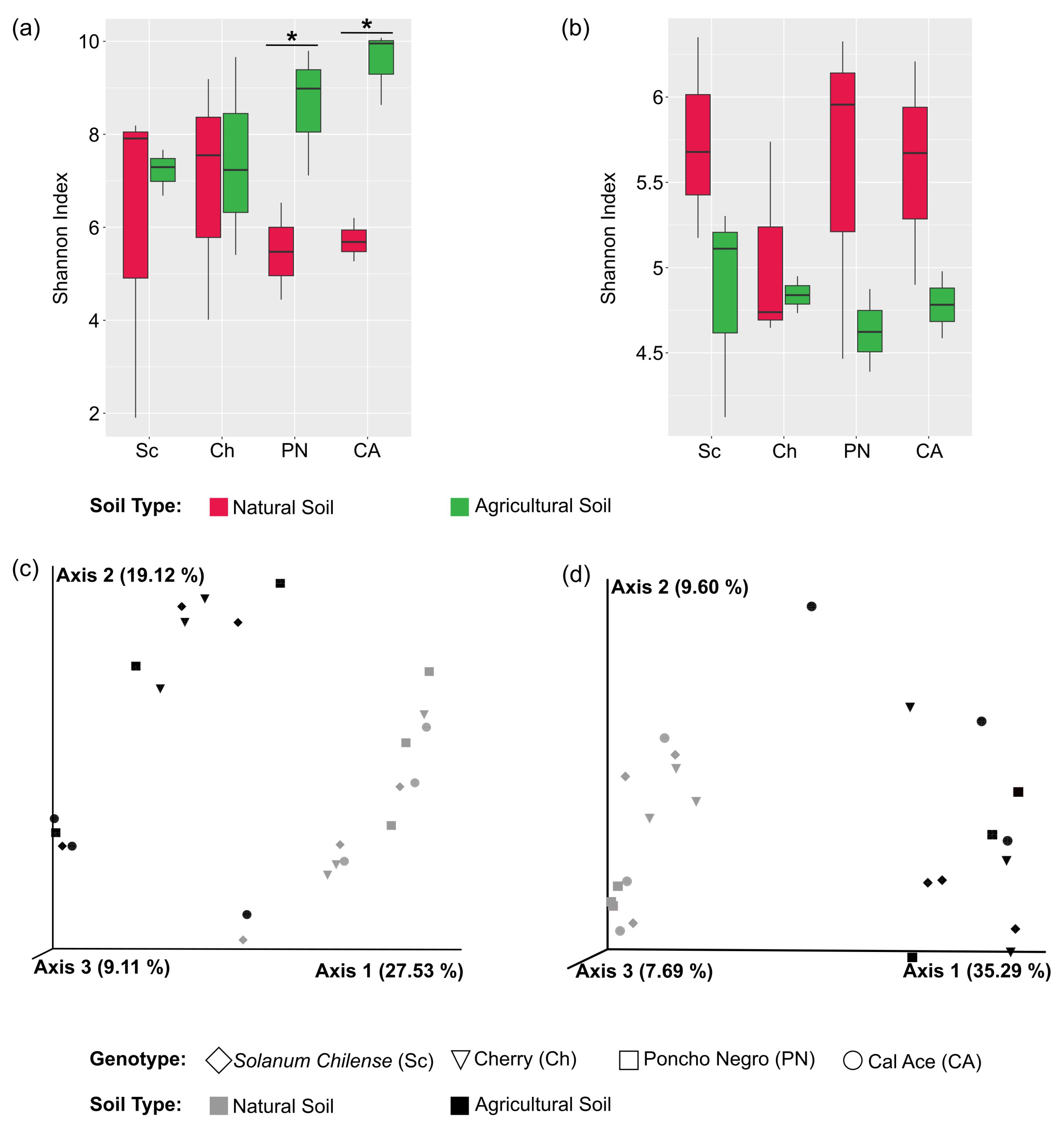
3.2.2. Alpha and Beta Diversity
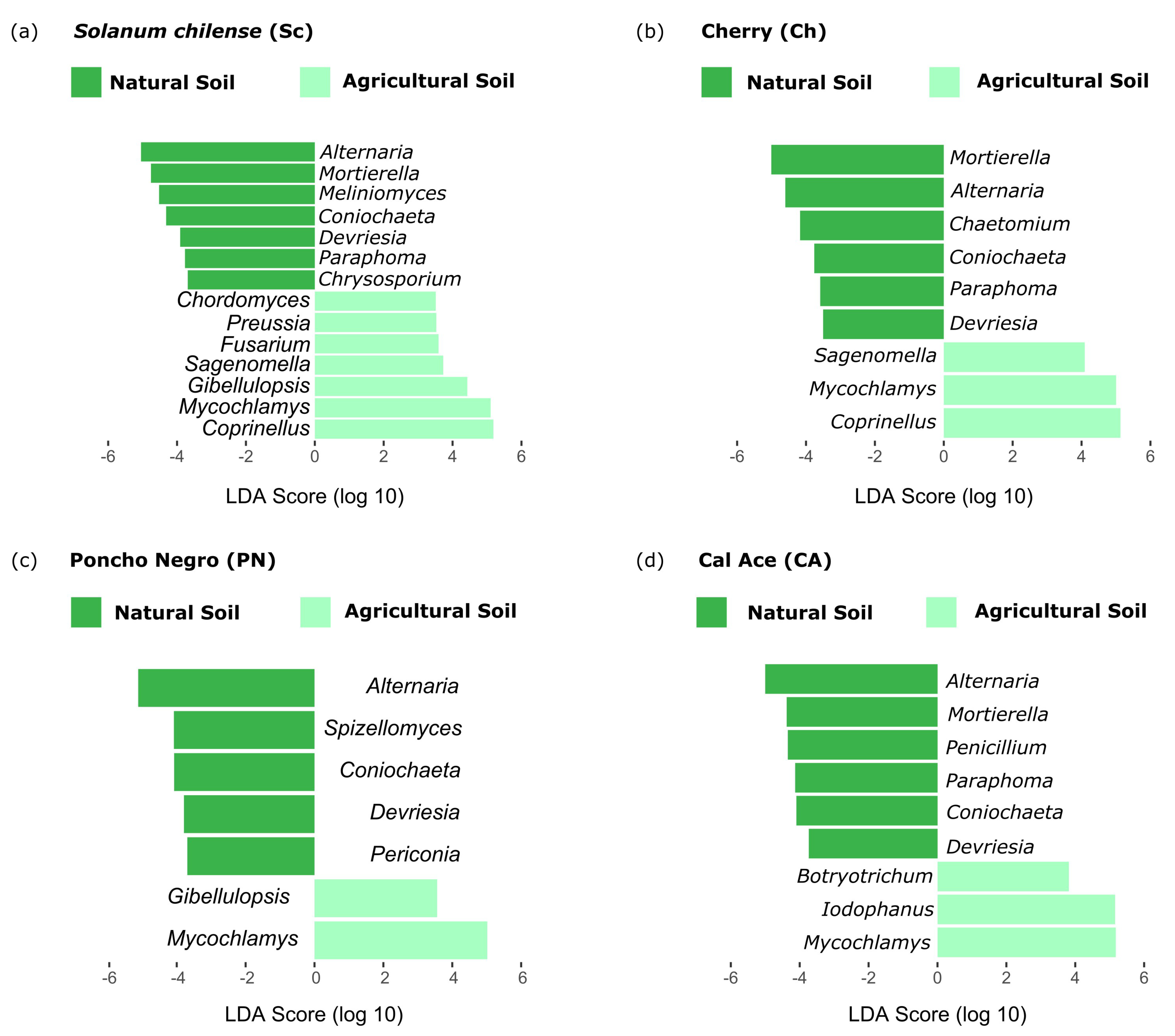
3.2.3. Effect of Tomato Genotype and Soil Type on Microbial Abundance
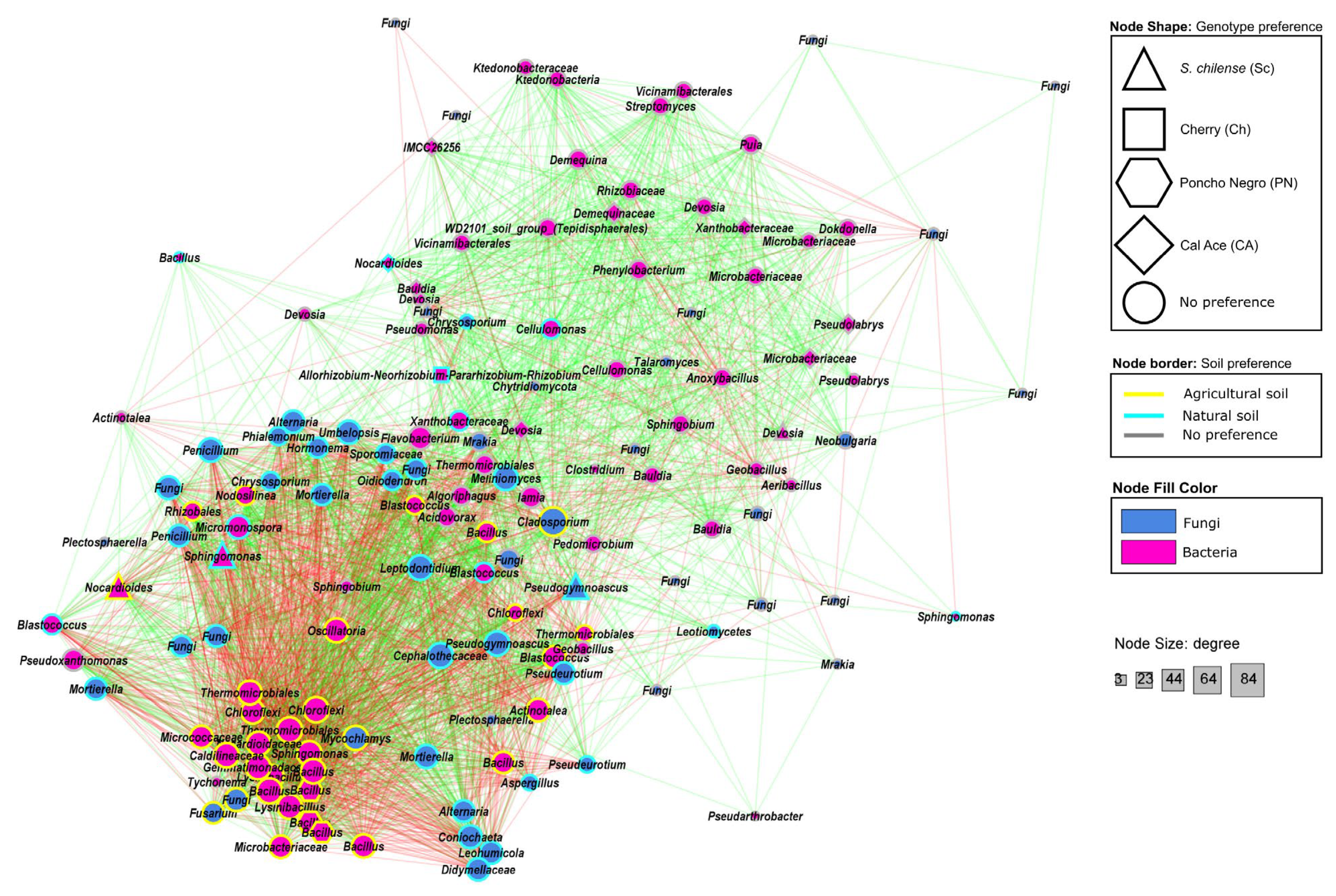
3.2.4. Co-Occurrence Patterns of Bacterial and Fungal Taxa
3.2.5. Functional Analysis of Bacterial Communities in the Tomato Rhizosphere
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oyserman, B.O.; Cordovez, V.; Flores, S.S.; Leite, M.F.A.; Nijveen, H.; Medema, M.H.; Raaijmakers, J.M. Extracting the GEMs: Genotype, Environment, and Microbiome Interactions Shaping Host Phenotypes. Front. Microbiol. 2021, 11, 574053. [Google Scholar] [CrossRef] [PubMed]
- Vives-Peris, V.; de Ollas, C.; Gómez-Cadenas, A.; Pérez-Clemente, R.M. Root Exudates: From Plant to Rhizosphere and Beyond. Plant Cell Rep. 2020, 39, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.; Foster, K.R. Host Control and the Evolution of Cooperation in Host Microbiomes. Nat. Commun. 2022, 13, 3567. [Google Scholar] [CrossRef] [PubMed]
- Rauf, S.; Silva, J. Da Consequences of Plant Breeding on Genetic Diversity. Int. J. Plant. Breed 2010, 4, 1–21. [Google Scholar]
- Begna, T. Effects of Crop Evolution under Domestication and Narrowing Genetic Bases of Crop Species. Open J. Plant Sci. 2021, 6, 49–54. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Mendes, R.; Raaijmakers, J.M. Impact of Plant Domestication on Rhizosphere Microbiome Assembly and Functions. Plant. Mol. Biol. 2016, 90, 635–644. [Google Scholar] [CrossRef]
- Yue, H.; Yue, W.; Jiao, S.; Kim, H.; Lee, Y.H.; Wei, G.; Song, W.; Shu, D. Plant Domestication Shapes Rhizosphere Microbiome Assembly and Metabolic Functions. Microbiome 2023, 11, 70. [Google Scholar] [CrossRef]
- Soldan, R.; Fusi, M.; Cardinale, M.; Daffonchio, D.; Preston, G.M. The Effect of Plant Domestication on Host Control of the Microbiota. Commun. Biol. 2021, 4, 936. [Google Scholar] [CrossRef]
- Hinsinger, P.; Bengough, A.G.; Vetterlein, D.; Young, I.M. Rhizosphere: Biophysics, Biogeochemistry and Ecological Relevance. Plant Soil 2009, 321, 117–152. [Google Scholar] [CrossRef]
- Santangeli, M.; Steininger-Mairinger, T.; Vetterlein, D.; Hann, S.; Oburger, E. Maize (Zea mays L.) Root Exudation Profiles Change in Quality and Quantity during Plant Development–A Field Study. Plant Sci. 2024, 338, 111896. [Google Scholar] [CrossRef]
- York, L.M.; Galindo-Castañeda, T.; Schussler, J.R.; Lynch, J.P. Evolution of US Maize (Zea mays L.) Root Architectural and Anatomical Phenes over the Past 100 Years Corresponds to Increased Tolerance of Nitrogen Stress. J. Exp. Bot. 2015, 66, 2347–2358. [Google Scholar] [CrossRef] [PubMed]
- Iannucci, A.; Fragasso, M.; Beleggia, R.; Nigro, F.; Papa, R. Evolution of the Crop Rhizosphere: Impact of Domestication on Root Exudates in Tetraploid Wheat (Triticum turgidum L.). Front. Plant Sci. 2017, 8, 2124. [Google Scholar] [CrossRef] [PubMed]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; Da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P.; et al. Dynamic Root Exudate Chemistry and Microbial Substrate Preferences Drive Patterns in Rhizosphere Microbial Community Assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Blanca, J.; Sanchez-Matarredona, D.; Ziarsolo, P.; Montero-Pau, J.; Van Der Knaap, E.; Díez, M.J.; Cañizares, J. Haplotype Analyses Reveal Novel Insights into Tomato History and Domestication Driven by Long-Distance Migrations and Latitudinal Adaptations. Hortic. Res. 2022, 9, uhac030. [Google Scholar] [CrossRef]
- Massaretto, I.L.; Albaladejo, I.; Purgatto, E.; Flores, F.B.; Plasencia, F.; Egea-Fernández, J.M.; Bolarin, M.C.; Egea, I. Recovering Tomato Landraces to Simultaneously Improve Fruit Yield and Nutritional Quality against Salt Stress. Front. Plant Sci. 2018, 9, 871. [Google Scholar] [CrossRef]
- Fullana-Pericàs, M.; Conesa, M.; Douthe, C.; El Aou-ouad, H.; Ribas-Carbó, M.; Galmés, J. Tomato Landraces as a Source to Minimize Yield Losses and Improve Fruit Quality under Water Deficit Conditions. Agric. Water Manag. 2019, 223, 105722. [Google Scholar] [CrossRef]
- Flores, S.S.; Cordovez, V.; Oyserman, B.; Stopnisek, N.; Raaijmakers, J.M.; van't Hof, P. The Tomato’s Tale: Exploring Taxonomy, Biogeography, Domestication, and Microbiome for Enhanced Resilience. Phytobiomes J. 2024, 8, 5–20. [Google Scholar] [CrossRef]
- Böndel, K.B.; Lainer, H.; Nosenko, T.; Mboup, M.; Tellier, A.; Stephan, W. North-South Colonization Associated with Local Adaptation of the Wild Tomato Species Solanum chilense. Mol. Biol. Evol. 2015, 32, 2932–2943. [Google Scholar] [CrossRef]
- Razifard, H.; Ramos, A.; Della Valle, A.L.; Bodary, C.; Goetz, E.; Manser, E.J.; Li, X.; Zhang, L.; Visa, S.; Tieman, D.; et al. Genomic Evidence for Complex Domestication History of the Cultivated Tomato in Latin America. Mol. Biol. Evol. 2020, 37, 1118–1132. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for de Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A.; StackCatchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An Analysis Tool Set for Population Genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Op De Beeck, M.; Lievens, B.; Busschaert, P.; Declerck, S.; Vangronsveld, J.; Colpaert, J.V. Comparison and Validation of Some ITS Primer Pairs Useful for Fungal Metabarcoding Studies. PLoS ONE 2014, 9, e97629. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids. Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; May, T.W.; Frøslev, T.G.; Pawlowska, J.; Lindahl, B.; Põldmaa, K.; Truong, C.; et al. The UNITE Database for Molecular Identificationãnd Taxonomic Communication of Fungiãnd Other Eukaryotes: Sequences, Taxaãnd Classifications r Econsider Ed. Nucleic Acids Res. 2024, 52, D791–D797. [Google Scholar] [CrossRef]
- Hall, M.; Beiko, R.G. 16S RRNA Gene Analysis with QIIME2. Methods Mol. Biol. 2018, 1849, 113–129. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and Qualitative β Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Wehrens, R.; Buydens, L. <Kohonen-Manual.Pdf>. JSS J. Stat. Softw. 2007, 21. [Google Scholar]
- Wickham, H. Programming with ggplot2. In ggplot2. Use R! Springer: Cham, Switzerland, 2016; pp. 241–253. [Google Scholar] [CrossRef]
- Cao, Y.; Dong, Q.; Wang, D.; Zhang, P.; Liu, Y.; Niu, C. MicrobiomeMarker: An R/Bioconductor Package for Microbiome Marker Identification and Visualization. Bioinformatics 2022, 38, 4027–4029. [Google Scholar] [CrossRef] [PubMed]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling Function and Taxonomy in the Global Ocean Microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T. Tax4Fun2: Prediction of Habitat-Specific Functional Profiles and Functional Redundancy Based on 16S RRNA Gene Sequences. Environ. Microbiome 2020, 7, 11. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. CoNet App: Inference of Biological Association Networks Using Cytoscape. F1000Res 2016, 5, 1519. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Ideker, T. Cytoscape: A Software Environment for Integrated Models. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kamada, T.; Kawai, S. An Algorithm for Drawing General Undirected Graphs. Inf. Process. Lett. 1989, 31, 7–15. [Google Scholar] [CrossRef]
- Kohl, M.; Wiese, S.; Warscheid, B. Chapter 18 and Analysis of Biological Networks. Methods 2011, 696, 291–303. [Google Scholar] [CrossRef]
- Anderson, M.J. A New Method for Non-Parametric Multivariate Analysis of Variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar]
- Oksanen, J.; Guillaume Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package, Version 2.6-2; Scientific Research Publishing Inc.: Irvine, CA, USA, 2022.
- R Core Team. R: A Language and Environment for Statistical Computing, Version 4.0.2; R Foundation for Statistical Computing: Vienna, Austria, 2020.
- Huang, J.; Li, Y.-F.; Ma, Y.-Y.; Li, Y.-S.; Jin, J.; Lian, T.-X. The Rhizospheric Microbiome Becomes More Diverse with Maize Domestication and Genetic Improvement. J. Integr. Agric. 2022, 21, 1188–1202. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Carrión, V.J.; Bosse, M.; Ferrão, L.F.V.; De Hollander, M.; Garcia, A.A.F.; Ramírez, C.A.; Mendes, R.; Raaijmakers, J.M. Linking Rhizosphere Microbiome Composition of Wild and Domesticated Phaseolus Vulgaris to Genotypic and Root Phenotypic Traits. ISME J. 2017, 11, 2244–2257. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Jaramillo, J.E.; De Hollander, M.; Ramírez, C.A.; Mendes, R.; Raaijmakers, J.M.; Carrión, V.J. Deciphering Rhizosphere Microbiome Assembly of Wild and Modern Common Bean (Phaseolus vulgaris) in Native and Agricultural Soils from Colombia. Microbiome 2019, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Mutch, L.A.; Young, J.P.W. Diversity and Specificity of Rhizobium Leguminosarum Biovar Viciae on Wild and Cultivated Legumes. Mol. Ecol. 2004, 13, 2435–2444. [Google Scholar] [CrossRef]
- Bouffaud, M.L.; Poirier, M.A.; Muller, D.; Moënne-Loccoz, Y. Root Microbiome Relates to Plant Host Evolution in Maize and Other Poaceae. Environ. Microbiol. 2014, 16, 2804–2814. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and Function of the Bacterial Root Microbiota in Wild and Domesticated Barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef]
- Simonin, M.; Dasilva, C.; Terzi, V.; Ngonkeu, E.L.M.; DIouf, D.; Kane, A.; Béna, G.; Moulin, L. Influence of Plant Genotype and Soil on the Wheat Rhizosphere Microbiome: Evidences for a Core Microbiome across Eight African and European Soils. FEMS Microbiol. Ecol. 2020, 96, fiaa067. [Google Scholar] [CrossRef]
- Liu, F.; Hewezi, T.; Lebeis, S.L.; Pantalone, V.; Grewal, P.S.; Staton, M.E. Soil Indigenous Microbiome and Plant Genotypes Cooperatively Modify Soybean Rhizosphere Microbiome Assembly. BMC Microbiol. 2019, 19, 201. [Google Scholar] [CrossRef]
- Wahab, A.; Muhammad, M.; Munir, A.; Abdi, G.; Zaman, W.; Ayaz, A.; Khizar, C.; Reddy, S.P.P. Role of Arbuscular Mycorrhizal Fungi in Regulating Growth, Enhancing Productivity, and Potentially Influencing Ecosystems under Abiotic and Biotic Stresses. Plants 2023, 12, 3102. [Google Scholar] [CrossRef]
- Oyserman, B.O.; Flores, S.S.; Griffioen, T.; Pan, X.; van der Wijk, E.; Pronk, L.; Lokhorst, W.; Nurfikari, A.; Paulson, J.N.; Movassagh, M.; et al. Disentangling the Genetic Basis of Rhizosphere Microbiome Assembly in Tomato. Nat. Commun. 2022, 13, 3228. [Google Scholar] [CrossRef]
- D’Angelo, T.; Goordial, J.; Lindsay, M.R.; McGonigle, J.; Booker, A.; Moser, D.; Stepanauskus, R.; Orcutt, B.N. Replicated Life-History Patterns and Subsurface Origins of the Bacterial Sister Phyla Nitrospirota and Nitrospinota. ISME J. 2023, 17, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Janarthine, S.R.S.; Eganathan, P. Plant Growth Promoting of Endophytic Sporosarcina Aquimarina SjAM16103 Isolated from the Pneumatophores of Avicennia Marina L. Int. J. Microbiol. 2012, 2012, 532060. [Google Scholar] [CrossRef]
- Sepúlveda Chavera, G.F.; Macuer, M.A.; Torres, P.M. Endospore-Forming Bacteria Present in a Commercial Stabilized Poultry Manure Determines the Fusarium Biocontrol and the Tomato Growth Promotion. Agronomy 2020, 10, 1636. [Google Scholar] [CrossRef]
- Seo, H.; Kim, J.H.; Lee, S.M.; Lee, S.W. The Plant-Associated Flavobacterium: A Hidden Helper for Improving Plant Health. Plant Pathol. J. 2024, 40, 251–260. [Google Scholar] [CrossRef]
- Guo, J.; Ling, N.; Li, Y.; Li, K.; Ning, H.; Shen, Q.; Guo, S.; Vandenkoornhuyse, P. Seed-Borne, Endospheric and Rhizospheric Core Microbiota as Predictors of Plant Functional Traits across Rice Cultivars Are Dominated by Deterministic Processes. New Phytol. 2021, 230, 2047–2060. [Google Scholar] [CrossRef]
- Gomes, W.d.S.; Partelli, F.L.; Veloso, T.G.R.; da Silva, M.d.C.S.; Moreli, A.P.; Moreira, T.R.; Pereira, L.L. Effects of Coffea Canephora Genotypes on the Microbial Community of Soil and Fruit. Sci. Rep. 2024, 14, 29035. [Google Scholar] [CrossRef]
- Saleem, M.; Hu, J.; Jousset, A. More Than the Sum of Its Parts: Microbiome Biodiversity as a Driver of Plant Growth and Soil Health. Annu. Rev. Ecol. Evol. Syst. 2019, 50, 145–168. [Google Scholar] [CrossRef]
- Wipf, H.M.-L.; Xu, L.; Gao, C.; Spinner, H.B.; Taylor, J.; Lemaux, P.; Mitchell, J.; Coleman-Derr, D. Agricultural Soil Management Practices Differentially Shape the Bacterial and Fungal Microbiomes of Sorghum Bicolor. Appl. Environ. Microbiol. 2021, 87, e02345-20. [Google Scholar] [CrossRef]
- Lori, M.; Leitao, R.; Felix, D.; Imbert, C.; Corti, A.; Cunha, L.; Symanczik, S.; Bünemann, E.; Creamer, R.; Vazquez, C. Response of Soil Biota to Agricultural Management Practices: A Systematic Quantitative Meta-Data-Analysis and Method Selection Framework. Soil Biol. Biochem. 2025, 207, 109815. [Google Scholar] [CrossRef]
- Hartman, K.; van der Heijden, M.G.A.; Wittwer, R.A.; Banerjee, S.; Walser, J.C.; Schlaeppi, K. Cropping Practices Manipulate Abundance Patterns of Root and Soil Microbiome Members Paving the Way to Smart Farming. Microbiome 2018, 6, 14. [Google Scholar] [CrossRef]
- Brisson, V.L.; Schmidt, J.E.; Northen, T.R.; Vogel, J.P.; Gaudin, A.C.M. Impacts of Maize Domestication and Breeding on Rhizosphere Microbial Community Recruitment from a Nutrient Depleted Agricultural Soil. Sci. Rep. 2019, 9, 15611. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Leung, P.M.; Wood, J.L.; Bay, S.K.; Hugenholtz, P.; Kessler, A.J.; Shelley, G.; Waite, D.W.; Franks, A.E.; Cook, P.L.M.; et al. Metabolic Flexibility Allows Bacterial Habitat Generalists to Become Dominant in a Frequently Disturbed Ecosystem. ISME J. 2021, 15, 2986–3004. [Google Scholar] [CrossRef] [PubMed]
- Hannula, S.E.; Di Lonardo, D.P.; Christensen, B.T.; Crotty, F.V.; Elsen, A.; van Erp, P.J.; Hansen, E.M.; Rubæk, G.H.; Tits, M.; Toth, Z.; et al. Inconsistent Effects of Agricultural Practices on Soil Fungal Communities across 12 European Long-Term Experiments. Eur. J. Soil Sci. 2021, 72, 1902–1923. [Google Scholar] [CrossRef]
- Wang, J.L.; Liu, K.L.; Zhao, X.Q.; Gao, G.F.; Wu, Y.H.; Shen, R.F. Microbial Keystone Taxa Drive Crop Productivity through Shifting Aboveground-Belowground Mineral Element Flows. Sci. Total Environ. 2022, 811, 152342. [Google Scholar] [CrossRef]
- Wang, C.; Kuzyakov, Y. Soil Organic Matter Priming: The PH Effects. Glob. Change Biol. 2024, 30, e17349. [Google Scholar] [CrossRef]
- Wu, C.; Yan, B.; Wei, F.; Wang, H.; Gao, L.; Ma, H.; Liu, Q.; Liu, Y.; Liu, G.; Wang, G. Long-Term Application of Nitrogen and Phosphorus Fertilizers Changes the Process of Community Construction by Affecting Keystone Species of Crop Rhizosphere Microorganisms. Sci. Total Environ. 2023, 897, 165239. [Google Scholar] [CrossRef]
- Smulders, L.; Benítez, E.; Moreno, B.; López-garcía, Á.; Pozo, M.J.; Ferrero, V.; Peña, E.d.l.; Alcalá Herrera, R. Tomato Domestication Affects Potential Functional Molecular Pathways of Root-associated Soil Bacteria. Plants 2021, 10, 1942. [Google Scholar] [CrossRef]
- Fang, J.; Blaschkauer, M.; Distelfeld, A.; Liu, Z.; Song, B.; Rachmilevitch, S.; Adams, J.M. Comparison of Rhizosphere Microbiomes Between Domesticated and Wild Wheat in a Typical Agricultural Field: Insights into Microbial Community Structure and Functional Shifts. J. Fungi 2025, 11, 168. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, M.; Araya-Angel, J.P.; Muñoz, A.; Alfaro-Flores, A.; Cardinale, M.; Stoll, A. Genetic Diversification of Tomato and Agricultural Soil Management Shaped the Rhizospheric Microbiome of Tomato (Solanum lycopersicum). Microorganisms 2025, 13, 1550. https://doi.org/10.3390/microorganisms13071550
González M, Araya-Angel JP, Muñoz A, Alfaro-Flores A, Cardinale M, Stoll A. Genetic Diversification of Tomato and Agricultural Soil Management Shaped the Rhizospheric Microbiome of Tomato (Solanum lycopersicum). Microorganisms. 2025; 13(7):1550. https://doi.org/10.3390/microorganisms13071550
Chicago/Turabian StyleGonzález, Máximo, Juan Pablo Araya-Angel, Ashlie Muñoz, Adalid Alfaro-Flores, Massimiliano Cardinale, and Alexandra Stoll. 2025. "Genetic Diversification of Tomato and Agricultural Soil Management Shaped the Rhizospheric Microbiome of Tomato (Solanum lycopersicum)" Microorganisms 13, no. 7: 1550. https://doi.org/10.3390/microorganisms13071550
APA StyleGonzález, M., Araya-Angel, J. P., Muñoz, A., Alfaro-Flores, A., Cardinale, M., & Stoll, A. (2025). Genetic Diversification of Tomato and Agricultural Soil Management Shaped the Rhizospheric Microbiome of Tomato (Solanum lycopersicum). Microorganisms, 13(7), 1550. https://doi.org/10.3390/microorganisms13071550