
The Abundance and Distribution of the acdS Gene in Microbial Communities from the Rhizosphere of Copiapoa solaris, a Native Cactus in the Arid Coastal Region of Antofagasta, Chile


Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Sites
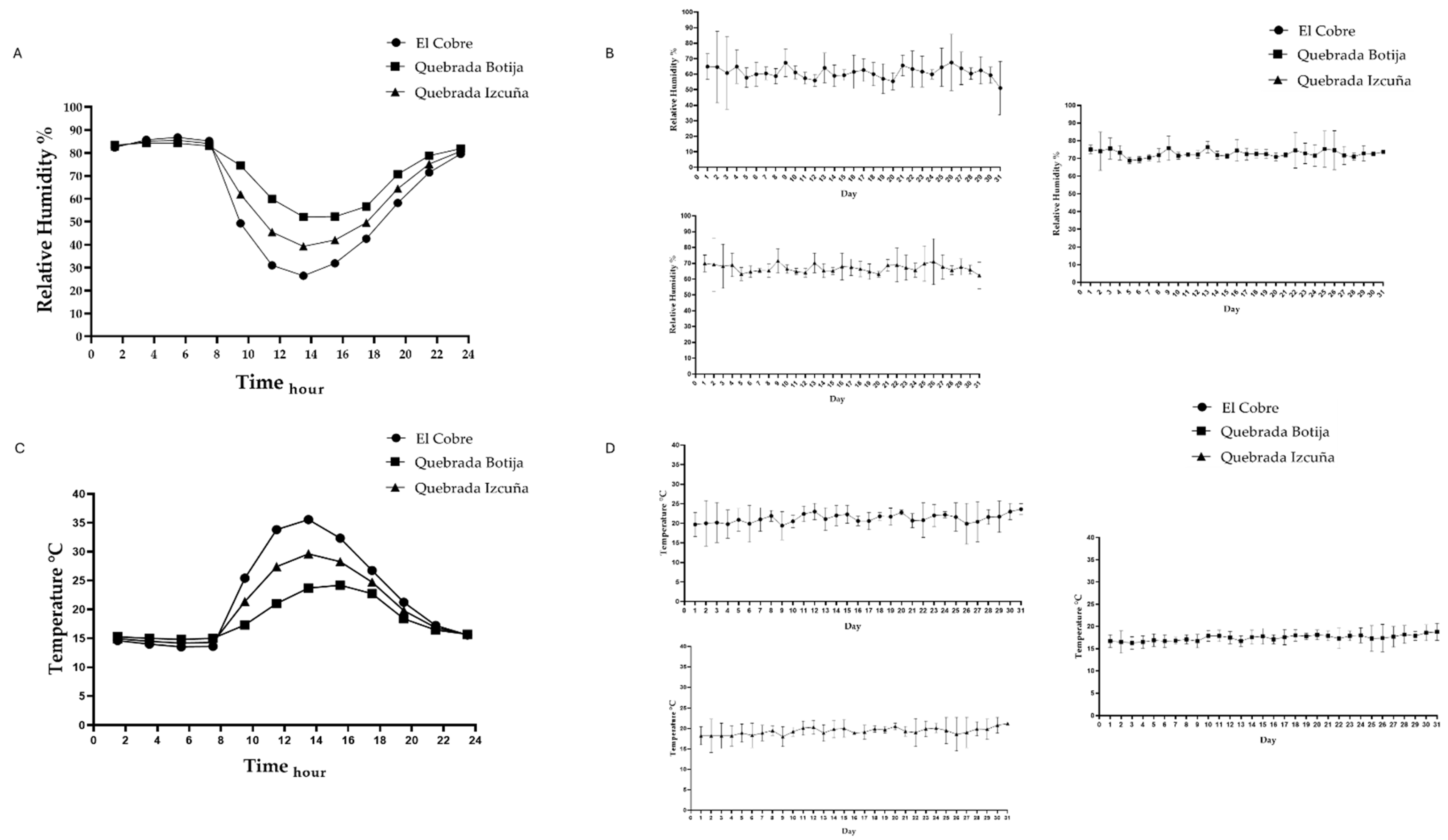
2.2. Environments Parameters
2.3. Environmental DNA Extraction and Shotgun Sequencing
2.4. Assembly and Taxonomy
2.5. acdS Gene Annotation
2.6. Phylogenetic Analysis
2.7. Mobile Element Reconstruction
2.8. Principal Component Analysis (PCA) Correlation
3. Results
3.1. Description Sampling Sites
3.2. Environmental DNA Extraction and Shotgun Sequencing
3.3. Assemblies and Taxonomy
3.4. acdS Gene Annotation
3.5. Phylogenetic Analysis
3.6. Mobile Element Reconstruction
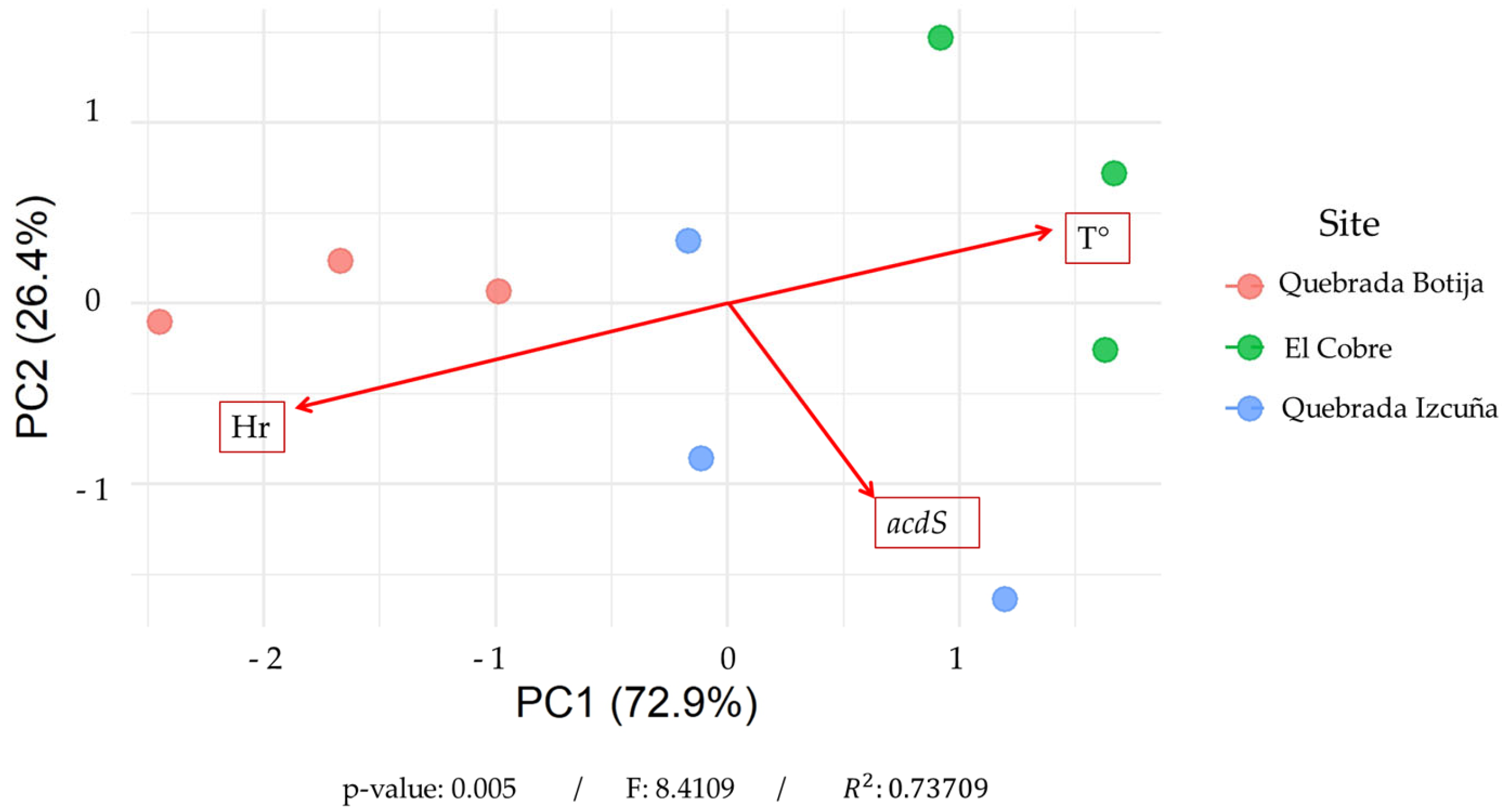
3.7. Principal Component Analysis (PCA) Correlation
4. Discussion
4.1. Environmental Influence on Microbial Diversity in the Rhizosphere of Copiapoa solaris
4.2. Taxonomic Composition and Functional Implications
4.3. Distribution and Mobility of the acdS Gene
4.4. Horizontal Gene Transfer and Metagenomic Assembly
4.5. Principal Component Analysis and Environmental Correlation
4.6. Implications for Conservation and Biotechnological Applications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Inglezakis, V.J.; Poulopoulos, S.G.; Arkhangelsky, E.; Zorpas, A.A.; Menegaki, A.N. Aquatic Environment. In Environment and Development: Basic Principles, Human Activities, and Environmental Implications; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 137–212. [Google Scholar] [CrossRef]
- Noy-Meir, I. Desert Ecosystems: Environment and Producers. Annu. Rev. Ecol. Syst. 1973, 4, 25–51. [Google Scholar] [CrossRef]
- Domen, J.K.; Stringfellow, W.T.; Camarillo, M.K.; Gulati, S. Fog water as an alternative and sustainable water resource. Clean Technol. Environ. Policy 2014, 16, 235–249. [Google Scholar] [CrossRef]
- Verbrugghe, N.; Khan, A.Z. Atmospheric water harvesting as a sustainable and resilient resource in arid climates: Gaining insights from ancient techniques. Water Supply 2024, 24, 3810–3830. [Google Scholar] [CrossRef]
- Schulz, R. Copiapoa; Schulz Publishing: Teesdale, Australia, 2006. [Google Scholar]
- Kavamura, V.N.; Santos, S.N.; da Silva, J.L.; Parma, M.M.; Ávila, L.A.; Visconti, A.; Zucchi, T.D.; Taketani, R.G.; Andreote, F.D.; de Melo, I.S. Screening of Brazilian cacti rhizobacteria for plant growth promotion under drought. Microbiol. Res. 2013, 168, 183–191. [Google Scholar] [CrossRef]
- de la Torre-Hernández, M.E.; Salinas-Virgen, L.I.; Aguirre-Garrido, J.F.; Fernández-González, A.J.; Martínez-Abarca, F.; Montiel-Lugo, D.; Ramírez-Saad, H.C. Composition, Structure, and PGPR Traits of the Rhizospheric Bacterial Communities Associated With Wild and Cultivated Echinocactus platyacanthus and Neobuxbaumia polylopha. Front. Microbiol. 2020, 11, 1424. [Google Scholar] [CrossRef]
- Riyazuddin, R.; Verma, R.; Singh, K.; Nisha, N.; Keisham, M.; Bhati, K.K.; Kim, S.T.; Gupta, R. Ethylene: A Master Regulator of Salinity Stress Tolerance in Plants. Biomolecules 2020, 10, 959. [Google Scholar] [CrossRef]
- Tao, J.J.; Chen, H.W.; Ma, B.; Zhang, W.K.; Chen, S.Y.; Zhang, J.S. The Role of Ethylene in Plants Under Salinity Stress. Front. Plant Sci. 2015, 27, 1059. [Google Scholar] [CrossRef]
- Chen, H.; Bullock, D.A.; Alonso, J.M.; Stepanova, A.N. To Fight or to Grow: The Balancing Role of Ethylene in Plant Abiotic Stress Responses. Plants 2022, 11, 33. [Google Scholar] [CrossRef]
- Gamalero, E.; Lingua, G.; Glick, B.R.; Ethylene, A.C.C. Plant Growth-Promoting Enzyme ACC Deaminase. Biology 2023, 12, 1043. [Google Scholar] [CrossRef]
- Singh, R.P.; Shelke, G.M.; Kumar, A.; Jha, P.N. Biochemistry and genetics of ACC deaminase: A weapon to “stress ethylene” produced in plants. Front. Microbiol. 2015, 6, 937. [Google Scholar] [CrossRef]
- Hontzeas, N.; Richardson, A.O.; Belimov, A.; Safronova, V.; Abu-Omar, M.M.; Glick, B.R. Evidence for Horizontal Transfer of 1-Aminocyclopropane-1-Carboxylate Deaminase Genes. Appl. Environ. Microbiol. 2005, 71, 7556–7558. [Google Scholar] [CrossRef] [PubMed]
- Schierstaedt, J.; Bziuk, N.; Kuzmanović, N.; Blau, K.; Smalla, K.; Jechalke, S. Role of Plasmids in Plant-Bacteria Interactions. Curr. Issues Mol. Biol. 2019, 30, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Manter, D.K.; Hamm, A.K.; Deel, H.L. Community structure and abundance of ACC deaminase containing bacteria in soils with 16S-PICRUSt2 inference or direct acdS gene sequencing. J. Microbiol. Methods 2023, 211, 106740. [Google Scholar] [CrossRef]
- Nascimento, F.X.; Rossi, M.J.; Soares, C.R.F.S.; McConkey, B.J.; Glick, B.R.; Dutilh, B.E. New Insights into 1-Aminocyclopropane-1-Carboxylate (ACC) Deaminase Phylogeny, Evolution and Ecological Significance. PLoS ONE 2014, 9, e99168. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Gupta, S. Diversity analysis of ACC deaminase producing bacteria associated with rhizosphere of coconut tree (Cocos nucifera L.) grown in Lakshadweep islands of India and their ability to promote plant growth under saline conditions. J. Biotechnol. 2020, 324, 183–197. [Google Scholar] [CrossRef]
- Bal, H.B.; Nayak, L.; Das, S.; Adhya, T.K. Isolation of ACC deaminase producing PGPR from rice rhizosphere and evaluating their plant growth promoting activity under salt stress. Plant Soil 2013, 366, 93–105. [Google Scholar] [CrossRef]
- Saraf, M.; Jha, C.K.; Patel, D. The Role of ACC Deaminase Producing PGPR in Sustainable Agriculture. In Plant Growth and Health Promoting Bacteria; Maheshwari, D.K., Ed.; Microbiology Monographs; Springer: Berlin/Heidelberg, Germany, 2010; Volume 18. [Google Scholar] [CrossRef]
- Singh, R.P.; Ma, Y.; Shadan, A. Perspective of ACC-deaminase producing bacteria in stress agriculture. J. Biotechnol. 2022, 352, 36–46. [Google Scholar] [CrossRef]
- Fuentes, B.; Gómez, F.; Valdez, C.; Videla, A.; Castro-Severyn, J.; Barahona, S.; Bol, R.; Riquelme, R.; Quispe, J.; Remonsellez, F. Effects of altitude on soil properties in coastal fog ecosystems in Morro Moreno National Park, Antofagasta, Chile. Eur. J. Soil. Sci. 2022, 73, e13217. [Google Scholar] [CrossRef]
- Barillot, C.D.C.; Sarde, C.-O.; Bert, V.; Tarnaud, E.; Cochet, N. A standardized method for the sampling of rhizosphere and rhizoplan soil bacteria associated to a herbaceous root system. Ann. Microbiol. 2013, 63, 471–476. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Van Goethem, M.W.; Pierneef, R.; Bezuidt, O.K.I.; Van De Peer, Y.; Cowan, D.A.; Makhalanyane, T.P. A reservoir of ‘historical’ antibiotic resistance genes in remote pristine Antarctic soils. Microbiome 2018, 6, 40. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Tang, X.; Shang, J.; Ji, Y.; Sun, Y. PLASMe: A tool to identify PLASMid contigs from short-read assemblies using transformer. Nucleic Acids Res. 2023, 51, e83. [Google Scholar] [CrossRef]
- da Costa, P.B.; Granada, C.E.; Ambrosini, A.; Moreira, F.; de Souza, R.; dos Passos, J.F.M.; Arruda, L.; Passaglia, L.M.P. A Model to Explain Plant Growth Promotion Traits: A Multivariate Analysis of 2,211 Bacterial Isolates. PLoS ONE 2014, 9, e116020. [Google Scholar] [CrossRef] [PubMed]
- Lado, C.; de Basanta, D.W.; Estrada-Torres, A.; Stephenson, S.L. The biodiversity of myxomycetes in central Chile. Fungal Divers. 2013, 59, 3–32. [Google Scholar] [CrossRef]
- Burger, S. Copiapoa Ecology: Pollinators, Parasites, Necrosis and a Mutualism with a Recently Described Ectinogonia Species. Cactus Succul. J. 2023, 95, 248–271. [Google Scholar] [CrossRef]
- Xiong, R.; Chen, Y. Molecular mechanisms and nutrient regulation of crop root responses to drought stress: Interactions with rhizosphere microorganisms. In Sustainable Agriculture under Drought Stress; Elsevier: Amsterdam, The Netherlands, 2025; pp. 499–509. [Google Scholar] [CrossRef]
- Bogati, K.; Walczak, M. The Impact of Drought Stress on Soil Microbial Community, Enzyme Activities and Plants. Agronomy 2022, 12, 189. [Google Scholar] [CrossRef]
- Dubey, A.; Malla, M.A.; Kumar, A.; Khan, M.L.; Kumari, S. Seed bio-priming with ACC deaminase-producing bacterial strains alleviates impact of drought stress in Soybean (Glycine max (L.) Merr.). Rhizosphere 2024, 30, 100873. [Google Scholar] [CrossRef]
- Farooq, M.; Hussain, M.; Wahid, A. Siddique KHM. Drought Stress in Plants: An Overview. In Plant Responses to Drought Stress; Aroca, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 1–33. [Google Scholar] [CrossRef]
- Şimşek, Ö.; Isak, M.A.; Dönmez, D.; Şekerci, A.D.; İzgü, T.; Kaçar, Y.A. Advanced Biotechnological Interventions in Mitigating Drought Stress in Plants. Plants 2024, 13, 717. [Google Scholar] [CrossRef]
- Buragohain, K.; Tamuly, D.; Sonowal, S.; Nath, R. Impact of Drought Stress on Plant Growth and Its Management Using Plant Growth Promoting Rhizobacteria. Indian J. Microbiol. 2024, 64, 287–303. [Google Scholar] [CrossRef]
- Ihsan, M.Z.; El-Nakhlawy, F.S.; Ismail, S.M.; Fahad, S.; Daur, I. Wheat Phenological Development and Growth Studies As Affected by Drought and Late Season High Temperature Stress under Arid Environment. Front. Plant Sci. 2016, 7, 795. [Google Scholar] [CrossRef]
- Rajanna, G.A.; Suman, A.; Venkatesh, P. Mitigating Drought Stress Effects in Arid and Semi-Arid Agro-Ecosystems through Bioirrigation Strategies—A Review. Sustainability 2023, 15, 3542. [Google Scholar] [CrossRef]
- Grayston, S.J.; Wang, S.; Campbell, C.D.; Edwards, A.C. Selective influence of plant species on microbial diversity in the rhizosphere. Soil. Biol. Biochem. 1998, 30, 369–378. [Google Scholar] [CrossRef]
- Mukhtar, S.; Mehnaz, S.; Malik, K.A. Microbial diversity in the rhizosphere of plants growing under extreme environments and its impact on crop improvement. Environ. Sustain. 2019, 2, 329–338. [Google Scholar] [CrossRef]
- Sarkar, A.; Saha, M.; Meena, V.S. Plant Beneficial Rhizospheric Microbes (PBRMs): Prospects for Increasing Productivity and Sustaining the Resilience of Soil Fertility. In Agriculturally Important Microbes for Sustainable Agriculture; Meena, V.S., Mishra, P.K., Bisht, J.K., Pattanayak, A., Eds.; Springer: Singapore, 2017. [Google Scholar] [CrossRef]
- Liu, S.; He, F.; Kuzyakov, Y.; Xiao, H.; Hoang, D.T.T.; Pu, S.; Razavi, B.S. Nutrients in the rhizosphere: A meta-analysis of content, availability, and influencing factors. Sci. Total Environ. 2022, 826, 153908. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.K.; Akhtar, M.S.; Panwar, J. Rhizospheric Plant-Microbe Interactions: Key Factors to Soil Fertility and Plant Nutrition. In Plant Microbes Symbiosis: Applied Facets; Arora, N.K., Ed.; Springer: New Delhi, India, 2015; Volume 6, pp. 127–145. [Google Scholar] [CrossRef]
- Fuerst, J.A. The PVC superphylum: Exceptions to the bacterial definition? Antonie Van Leeuwenhoek 2013, 104, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Ma, J.B.; Huang, Y.; Muhammad, M.; Lian, H.T.; Shurigin, V.; Egamberdieva, D.; Li, W.J.; Li, L. Insight into endophytic microbial diversity in two halophytes and plant beneficial attributes of Bacillus swezeyi. Front. Microbiol. 2024, 15, 1447755. [Google Scholar] [CrossRef]
- Peng, Y.; He, X.; Tao, Y.; Zhou, C.; Li, X. The Changes of the Endophytic Bacterial Community from Pepper Varieties with Different Capsaicinoids. Microorganisms. 2025, 13, 596. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, H.; Liu, W.; Sun, J.; Zhao, M.; Han, G.; Pan, Q. Effects of grazing intensity on diversity and composition of rhizosphere and non-rhizosphere microbial communities in a desert grassland. Ecol. Evol. 2023, 13, e10300. [Google Scholar] [CrossRef]
- Handique, M.; Bora, P.; Ziogas, V.; Srivastava, A.K.; Jagannadham, P.T.K.; Das, A.K. Phytophthora Infection Reorients the Composition of Rhizospheric Microbial Assembly in Khasi Mandarin (Citrus reticulata Blanco). Agronomy 2024, 14, 661. [Google Scholar] [CrossRef]
- Yadav, A.N.; Verma, P.; Kumar, S.; Kumar, V.; Kumar, M.; Sugitha, T.C.K.; Singh, B.P.; Saxena, A.K.; Dhaliwal, H.S. Actinobacteria from Rhizosphere: Molecular Diversity, Distributions, and Potential Biotechnological Applications; Elsevier: Amsterdam, The Netherlands, 2018; pp. 13–41. [Google Scholar] [CrossRef]
- Lu, Y.; Rosencrantz, D.; Liesack, W.; Conrad, R. Structure and activity of bacterial community inhabiting rice roots and the rhizosphere. Environ. Microbiol. 2006, 8, 1351–1360. [Google Scholar] [CrossRef]
- Pan, X.; Raaijmakers, J.M.; Carrión, V.J. Importance of Bacteroidetes in host–microbe interactions and ecosystem functioning. Trends Microbiol. 2023, 31, 959–971. [Google Scholar] [CrossRef]
- Ling, N.; Wang, T.; Kuzyakov, Y. Rhizosphere bacteriome structure and functions. Nat. Commun. 2022, 13, 836. [Google Scholar] [CrossRef]
- Soussi, A.; Ferjani, R.; Marasco, R.; Guesmi, A.; Cherif, H.; Rolli, E.; Mapelli, F.; Ouzari, H.I.; Daffonchio, D.; Cherif, A. Plant-associated microbiomes in arid lands: Diversity, ecology and biotechnological potential. Plant Soil 2016, 405, 357–370. [Google Scholar] [CrossRef]
- Mohammadipanah, F.; Wink, J. Actinobacteria from Arid and Desert Habitats: Diversity and Biological Activity. Front. Microbiol. 2016, 6, 1541. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, X.; Zuo, Y.; Hu, Q.; He, X. Plant species shape the bacterial communities on the phyllosphere in a hyper-arid desert. Microbiol. Res. 2023, 269, 127314. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Nan, Q.; Liu, Y.; Dong, D.; Qin, Y.; Li, S.; Wu, W. Stress resistance enhancing with biochar application and promotion on crop growth. Biochar 2024, 6, 43. [Google Scholar] [CrossRef]
- Vurukonda, S.S.K.P.; Vardharajula, S.; Shrivastava, M.; SkZ, A. Enhancement of drought stress tolerance in crops by plant growth promoting rhizobacteria. Microbiol. Res. 2016, 184, 13–24. [Google Scholar] [CrossRef]
- Bargaz, A.; Lyamlouli, K.; Chtouki, M.; Zeroual, Y.; Dhiba, D. Soil Microbial Resources for Improving Fertilizers Efficiency in an Integrated Plant Nutrient Management System. Front. Microbiol. 2018, 9, 1606. [Google Scholar] [CrossRef]
- Richardson, A.E.; Barea, J.M.; McNeill, A.M.; Prigent-Combaret, C. Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms. Plant Soil 2009, 321, 305–339. [Google Scholar] [CrossRef]
- Liu, Y.; Wilson, A.J.; Han, J.; Hui, A.; O’sUllivan, L.; Huan, T.; Haney, C.H.; Guttman, D.S. Amino Acid Availability Determines Plant Immune Homeostasis in the Rhizosphere Microbiome. mBio 2023, 14, e0342422. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; Paulitz, T.C.; Steinberg, C.; Alabouvette, C.; Moënne-Loccoz, Y. The rhizosphere: A playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 2009, 321, 341–361. [Google Scholar] [CrossRef]
- de Faria, M.R.; Costa, L.S.A.S.; Chiaramonte, J.B.; Bettiol, W.; Mendes, R. The rhizosphere microbiome: Functions, dynamics, and role in plant protection. Trop. Plant Pathol. 2021, 46, 13–25. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. 2013, 37, 634–663. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, R.M.; Ford, B.L.; Li, L.; Aroney, S.T.N.; Knights, H.E.; Ledermann, R.; East, A.K.; Ramachandran, V.K.; Poole, P.S. Lifestyle adaptations of Rhizobium from rhizosphere to symbiosis. Proc. Natl. Acad. Sci. USA 2020, 117, 23823–23834. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Philp, J.; Wang, Y.; Hu, J.; Wei, Y.; Li, J.; Ryder, M.; Toh, R.; Zhou, Y.; Denton, M.D.; et al. Plant growth-promoting rhizobacteria Burkholderia vietnamiensis B418 inhibits root-knot nematode on watermelon by modifying the rhizosphere microbial community. Sci. Rep. 2022, 12, 8381. [Google Scholar] [CrossRef]
- Govindasamy, V.; Senthilkumar, M.; Mageshwaran, V.; Annapurna, K. Detection and Characterization of ACC Deaminase in Plant Growth Promoting Rhizobacteria. J. Plant Biochem. Biotechnol. 2009, 18, 71–76. [Google Scholar] [CrossRef]
- Mohammad, M.; Badaluddin, N.A.; Asri, E.A. Biological functions of Paenibacillus spp. for agriculture applicationst. Bulg. J. Agric. Sci. 2024, 30, 930–947. [Google Scholar]
- Omar, A.F.; Abdelmageed, A.H.A.; Al-Turki, A.; Abdelhameid, N.M.; Sayyed, R.Z.; Rehan, M. Exploring the Plant Growth-Promotion of Four Streptomyces Strains from Rhizosphere Soil to Enhance Cucumber Growth and Yield. Plants 2022, 11, 3316. [Google Scholar] [CrossRef]
- Hirota, Y.; Fujii, T.; Sano, Y.; Iyama, S. Nitrogen fixation in the rhizosphere of rice. Nature 1978, 276, 416–417. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, N.; Zhang, Z.; Lei, C.; Chen, B.; Qin, G.; Qiu, D.; Lu, T.; Qian, H. Deciphering Microbial Community and Nitrogen Fixation in the Legume Rhizosphere. J. Agric. Food Chem. 2024, 72, 5659–5670. [Google Scholar] [CrossRef]
- Quintas-Nunes, F.; Brandão, P.R.; Barreto Crespo, M.T.; Glick, B.R.; Nascimento, F.X. Plant Growth Promotion, Phytohormone Production and Genomics of the Rhizosphere-Associated Microalga, Micractinium rhizosphaerae sp. nov. Plants 2023, 12, 651. [Google Scholar] [CrossRef]
- Sokolova, M.G.; Akimova, G.P.; Vaishlya, O.B. Effect of phytohormones synthesized by rhizosphere bacteria on plants. Appl. Biochem. Microbiol. 2011, 47, 274–278. [Google Scholar] [CrossRef]
- Ma, B.; Zhou, Z.Y.; Zhang, C.P.; Zhang, G.; Hu, Y.J. Inorganic phosphorus fractions in the rhizosphere of xerophytic shrubs in the Alxa Desert. J. Arid. Environ. 2009, 73, 55–61. [Google Scholar] [CrossRef]
- Nourashrafeddin, S.M.; Ramandi, A.; Seifi, A. Rhizobacteria isolated from xerophyte Haloxylon ammodendron manipulate root system architecture and enhance drought and salt tolerance in Arabidopsis thaliana. Int. Microbiol. 2023, 27, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Albureikan, M.O.I. Rhizosphere Microorganisms with Different Strategies and Mechanisms to Enhance Plant Growth in the Occurrence of Different Environmental Stress Factors. J. Pure Appl. Microbiol. 2023, 17, 1341–1355. [Google Scholar] [CrossRef]
- Khare, T.; Jamla, M.; Mathur, V.; Kumar, V. Exploring Halobiome Resources for Developing Salt-Tolerant Crops: A Perspective Review. J. Plant Growth Regul. 2024, 43, 2137–2164. [Google Scholar] [CrossRef]
- Malambane, G.; Nonaka, S.; Shiba, H.; Ezura, H.; Tsujimoto, H.; Akashi, K. Comparative effects of ethylene inhibitors on Agrobacterium mediated transformation of drought-tolerant wild watermelon. Biosci. Biotechnol. Biochem. 2018, 82, 433–441. [Google Scholar] [CrossRef]
- Daunoras, J.; Kačergius, A.; Gudiukaitė, R. Role of Soil Microbiota Enzymes in Soil Health and Activity Changes Depending on Climate Change and the Type of Soil Ecosystem. Biology 2024, 13, 85. [Google Scholar] [CrossRef]
- Zheng, Y.; Tang, J.; Liu, C.; Liu, X.; Luo, Z.; Zou, D.; Xiang, G.; Bai, J.; Meng, G.; Liu, X.; et al. Alleviation of metal stress in rape seedlings (Brassica napus L.) using the antimony-resistant plant growth-promoting rhizobacteria Cupriavidus sp. S-8-2. Sci. Total Environ. 2023, 858, 159955. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, W.; Xiao, H.; Zhai, Y.; Luo, Y.; Wang, Y.; Liu, Z.; Li, Q.; Huang, J. A Review on Rhizosphere Microbiota of Tea Plant (Camellia sinensis L.): Recent Insights and Future Perspectives. J. Agric. Food Chem. 2023, 71, 19165–19188. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, X.; Ouyang, W.; Yang, E.; Cao, Y.; Sun, R. Lowered Cd toxicity, uptake and expression of metal transporter genes in maize plant by ACC deaminase-producing bacteria Achromobacter sp. J. Hazard. Mater. 2022, 423, 127036. [Google Scholar] [CrossRef]
- Danish, S.; Zafar-Ul-Hye, M.; Fahad, S.; Saud, S.; Brtnicky, M.; Hammerschmiedt, T.; Datta, R. Drought Stress Alleviation by ACC Deaminase Producing Achromobacter xylosoxidans and Enterobacter cloacae, with and without Timber Waste Biochar in Maize. Sustainability 2020, 12, 6286. [Google Scholar] [CrossRef]
- Rashad, E.M.; Shaheen, D.M.; Al-Askar, A.A.; Ghoneem, K.M.; Arishi, A.A.; Hassan, E.S.A.; Saber, W.I. Seed Endophytic Achromobacter sp. F23KW as a Promising Growth Promoter and Biocontrol of Rhizoctonia Root Rot of Fenugreek. Molecules 2022, 27, 5546. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Quan, S.; Li, L.; Lei, G.; Li, S.; Gong, T.; Zhang, Z.; Hu, Y.; Yang, W. Endophytic Bacteria Improve Bio- and Phytoremediation of Heavy Metals. Microorganisms 2024, 12, 2137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Q.; Gao, C.; Lu, Y.; Sheng, Y.; Xiao, M.; Yun, Y.; Selvaraj, J.N.; Zhang, X.; Li, Y.; et al. Endophytic bacteria for Cd remediation in rice: Unraveling the Cd tolerance mechanisms of Cupriavidus metallidurans CML2. J. Hazard. Mater. 2024, 469, 133846. [Google Scholar] [CrossRef]
- De Luca, G.; Barakat, M.; Verméglio, A.; Achouak, W.; Heulin, T. The Bacterial Genus Ramlibacter: Betaproteobacteria Capable of Surviving in Oligotrophic Environments Thanks to Several Shared Genetic Adaptation Traits. Environ. Microbiol. 2025, 27, e70059. [Google Scholar] [CrossRef]
- Schwinning, S.; Ehleringer, J.R. Water use trade-offs and optimal adaptations to pulse-driven arid ecosystems. J. Ecol. 2001, 89, 464–480. [Google Scholar] [CrossRef]
- Schimel, J.; Balser, T.C.; Wallenstein, M. microbial stress-response physiology and its implications for ecosystem function. Ecology 2007, 88, 1386–1394. [Google Scholar] [CrossRef]
- Kehlet-Delgado, H.; Montoya, A.P.; Jensen, K.T.; Wendlandt, C.E.; Dexheimer, C.; Roberts, M.; Martínez, L.T.; Friesen, M.L.; Griffitts, J.S.; Porter, S.S. The evolutionary genomics of adaptation to stress in wild rhizobium bacteria. Proc. Natl. Acad. Sci. USA 2024, 121, e2311127121. [Google Scholar] [CrossRef]
- Wang, M.; Wang, L.; Shi, H.; Liu, Y.; Chen, S. Soil bacteria, genes, and metabolites stimulated during sulfur cycling and cadmium mobilization under sodium sulfate stress. Environ. Res. 2021, 201, 111599. [Google Scholar] [CrossRef]
- Gureeva, M.V.; Gureev, A.P. Molecular Mechanisms Determining the Role of Bacteria from the Genus Azospirillum in Plant Adaptation to Damaging Environmental Factors. Int. J. Mol. Sci. 2023, 24, 9122. [Google Scholar] [CrossRef]
- Ku, Y.S.; Wang, Z.; Duan, S.; Lam, H.M. Rhizospheric Communication through Mobile Genetic Element Transfers for the Regulation of Microbe–Plant Interactions. Biology 2021, 10, 477. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.H.; Jeong, H.Y.; Jung, S.K.; Kim, C.K. Overexpression of 1-Aminocyclopropane-1-Carboxylic Acid Deaminase (acdS) Gene in Petunia hybrida Improves Tolerance to Abiotic Stresses. Front. Plant Sci. 2021, 12, 737490. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Chen, Y.; Chen, S.; Yan, D.; Wang, X.; Guo, Y. AcdS gene of Bacillus cereus enhances salt tolerance of seedlings in tobacco (Nicotiana tabacum L.). Biotechnol. Biotechnol. 2022, 36, 902–913. [Google Scholar] [CrossRef]
- Baek, S.; Naing, A.H.; Kang, H.; Chung, M.Y.; Kim, C.K. Overexpression of acdS in petunia reduces ethylene production and improves tolerance to heat stress. Plant Biol. 2024, 26, 789–797. [Google Scholar] [CrossRef]
- De Vries, F.T.; Shade, A. Controls on soil microbial community stability under climate change. Front. Microbiol. 2013, 4, 265. [Google Scholar] [CrossRef]
- Zhang, Y.; Tariq, A.; Hughes, A.C.; Hong, D.; Wei, F.; Sun, H.; Sardans, J.; Peñuelas, J.; Perry, G.; Qiao, J. Challenges and solutions to biodiversity conservation in arid lands. Sci. Total Environ. 2023, 857, 159695. [Google Scholar] [CrossRef]
- Villalobo-Lopez, A.; Peña, C.M.; Varas-Myrik, A.; Pillet, M.; Jahnsen, P.; Pliscoff, P.; Goettsch, B.; Guerrero, P.C. Effects of trade and poaching pressure on extinction risk for cacti in the Atacama Desert. Conserv. Biol. 2024, 38, e14353. [Google Scholar] [CrossRef]
- Ortega-Baes, P.; Sühring, S.; Sajama, J.; Sotola, E.; Alonso-Pedano, M.; Bravo, S.; Godínez-Alvarez, H. Diversity and Conservation in the Cactus Family; Springer: Berlin/Heidelberg, Germany, 2010; pp. 157–173. [Google Scholar] [CrossRef]
- Heydarian, Z.; Yu, M.; Gruber, M.; Glick, B.R.; Zhou, R.; Hegedus, D.D. Inoculation of Soil with Plant Growth Promoting Bacteria Producing 1-Aminocyclopropane-1-Carboxylate Deaminase or Expression of the Corresponding acdS Gene in Transgenic Plants Increases Salinity Tolerance in Camelina sativa. Front. Microbiol. 2016, 7, 1966. [Google Scholar] [CrossRef]
- Xu, D.; Yu, X.; Yang, J.; Zhao, X.; Bao, Y. High-Throughput Sequencing Reveals the Diversity and Community Structure in Rhizosphere Soils of Three Endangered Plants in Western Ordos, China. Curr. Microbiol. 2020, 77, 2713–2723. [Google Scholar] [CrossRef]
- Li, F.; Lu, S.; Sun, W. Evaluating the Endangered Mechanism and Different Conservation Effects of Pinus squamata from the Rhizosphere Soil Bacterial Community Perspective. Biol. Life Sci. 2024, 12, 638. [Google Scholar] [CrossRef]
- Mikiciuk, G.; Miller, T.; Kisiel, A.; Cembrowska-Lech, D.; Mikiciuk, M.; Łobodzińska, A.; Bokszczanin, K. Harnessing Beneficial Microbes for Drought Tolerance: Ecological and Agricultural Innovations. Agriculture 2024, 14, 2228. [Google Scholar] [CrossRef]
- Bittencourt, P.P.; Alves, A.F.; Ferreira, M.B.; Da Silva Irineu, L.E.S.; Pinto, V.B.; Olivares, F.L. Mechanisms and Applications of Bacterial Inoculants in Plant Drought Stress Tolerance. Microorganisms 2023, 11, 502. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Latitude/Longitude | |
|---|---|---|
| El Cobre | C1 | −24.2966 S/−70.4920 W |
| C2 | −24.3022 S/−70.4948 W | |
| C3 | −24.3048 S/−70.5058 W | |
| Quebrada Botija | B1 | −24.5007 S/−70.5329 W |
| B2 | −24.5017 S/−70.5325 W | |
| B3 | −24.5022 S/−70.5330 W | |
| Quebrada Izcuña | I1 | −24.4283 S/−70.5302 W |
| I2 | −24.4278 S/−70.5303 W | |
| I3 | −24.4279 S/−70.5298 W | |
| Samples | Number of Contigs | N50 | L50 |
|---|---|---|---|
| B1 | 118,985 contigs greater than 1000 bp | 1279 | 70,268 |
| B2 | 114,205 contigs greater than 1000 bp | 1250 | 70,654 |
| B3 | 119,063 contigs greater than 1000 bp | 1230 | 75,425 |
| C1 | 92,533 contigs greater than 1000 bp | 1100 | 70,565 |
| C2 | 55,944 contigs greater than 1000 bp | 876 | 74,112 |
| C3 | 60,780 contigs greater than 1000 bp | 966 | 63,022 |
| I1 | 112,580 contigs greater than 1000 bp | 1202 | 80,825 |
| I2 | 118,204 contigs greater than 1000 bp | 1092 | 93,283 |
| I3 | 61,411 contigs greater than 1000 bp | 991 | 59,346 |
| Samples | Number of Plasmids >800 |
|---|---|
| C1 | 2779 |
| C2 | 2558 |
| C3 | 2502 |
| B1 | 2788 |
| B2 | 2832 |
| B3 | 2832 |
| I1 | 4600 |
| I2 | 6121 |
| I3 | 2636 |
| Sample | % Identity | Coverage | Plasmid ID |
|---|---|---|---|
| C2 | 64% | 240 aa | K141_255461 |
| I1 | 76% | 204 aa | K141_550940 |
| I1 | 76% | 316 aa | K141_772727 |
| I2 | 60% | 311 aa | K141_684709 |
| I3 | 63% | 245 aa | K141_341888 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cayo, M.; Solís-Cornejo, F.; Santos, A.; Zamorano, P.; Valenzuela, B. The Abundance and Distribution of the acdS Gene in Microbial Communities from the Rhizosphere of Copiapoa solaris, a Native Cactus in the Arid Coastal Region of Antofagasta, Chile. Microorganisms 2025, 13, 1547. https://doi.org/10.3390/microorganisms13071547
Cayo M, Solís-Cornejo F, Santos A, Zamorano P, Valenzuela B. The Abundance and Distribution of the acdS Gene in Microbial Communities from the Rhizosphere of Copiapoa solaris, a Native Cactus in the Arid Coastal Region of Antofagasta, Chile. Microorganisms. 2025; 13(7):1547. https://doi.org/10.3390/microorganisms13071547
Chicago/Turabian StyleCayo, Mayra, Francisco Solís-Cornejo, Andrés Santos, Pedro Zamorano, and Bernardita Valenzuela. 2025. "The Abundance and Distribution of the acdS Gene in Microbial Communities from the Rhizosphere of Copiapoa solaris, a Native Cactus in the Arid Coastal Region of Antofagasta, Chile" Microorganisms 13, no. 7: 1547. https://doi.org/10.3390/microorganisms13071547
APA StyleCayo, M., Solís-Cornejo, F., Santos, A., Zamorano, P., & Valenzuela, B. (2025). The Abundance and Distribution of the acdS Gene in Microbial Communities from the Rhizosphere of Copiapoa solaris, a Native Cactus in the Arid Coastal Region of Antofagasta, Chile. Microorganisms, 13(7), 1547. https://doi.org/10.3390/microorganisms13071547