
Dynamics of Microorganisms and Metabolites in the Mixed Silage of Oats and Vetch in Alpine Pastures, and Their Regulatory Mechanisms Under Low Temperatures


Abstract
1. Introduction
2. Materials and Methods
2.1. Silage Preparation
2.2. Determination of Fermentation Parameters and Nutrient Content
2.3. Microbial Community Analysis
2.4. Metabolic Profile Analyses
2.5. Data Analysis
3. Results
3.1. Silage Fermentation Quality
3.2. Chemical Composition of Mixed Silage at 0 Days and 90 Days
3.3. Bacterial Community Composition and Diversity of Mixed Silage at 0 Days and 90 Days
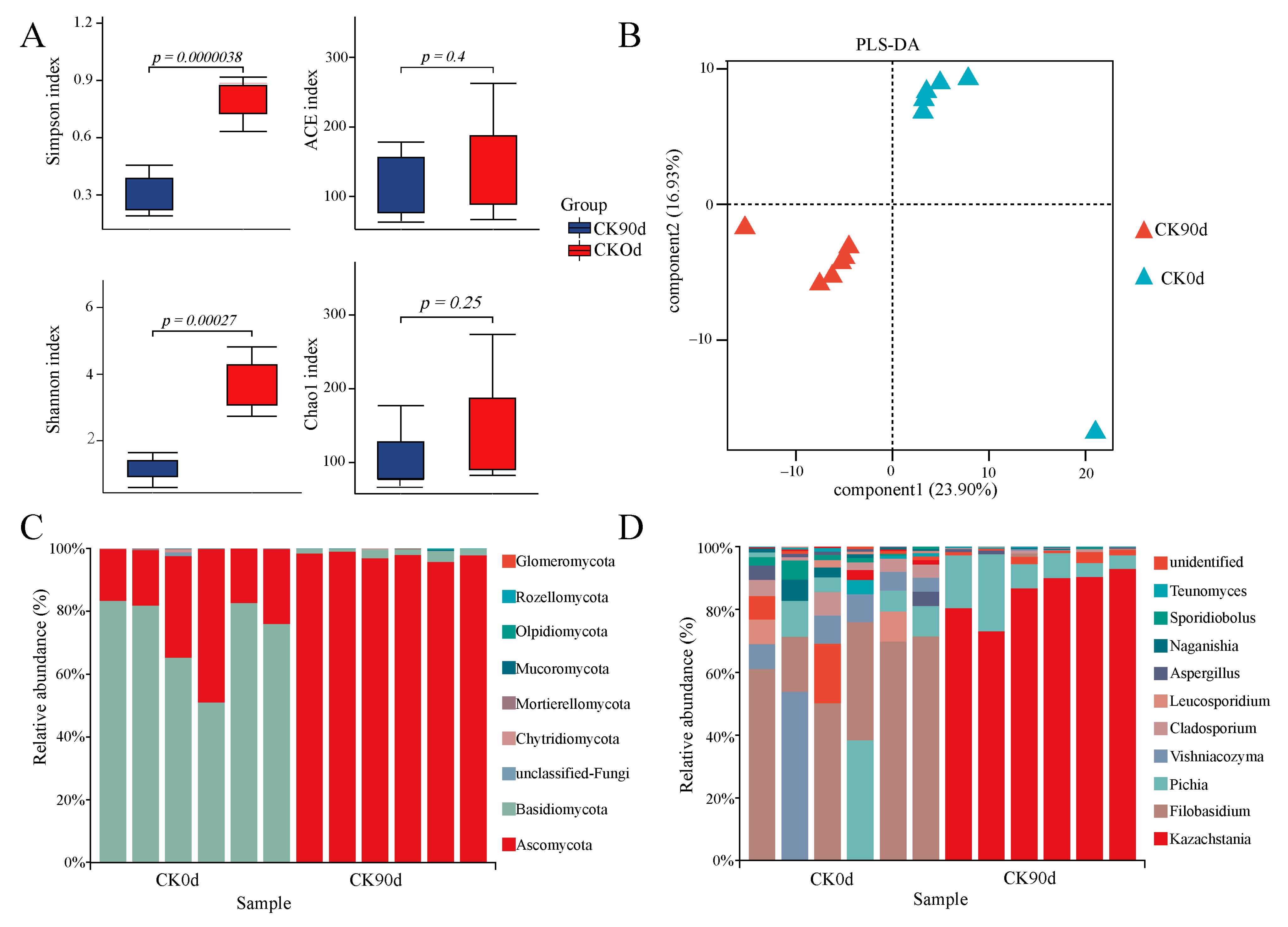
3.4. Fungal Community Composition and Diversity of Mixed Silage at 0 Days and 90 Days
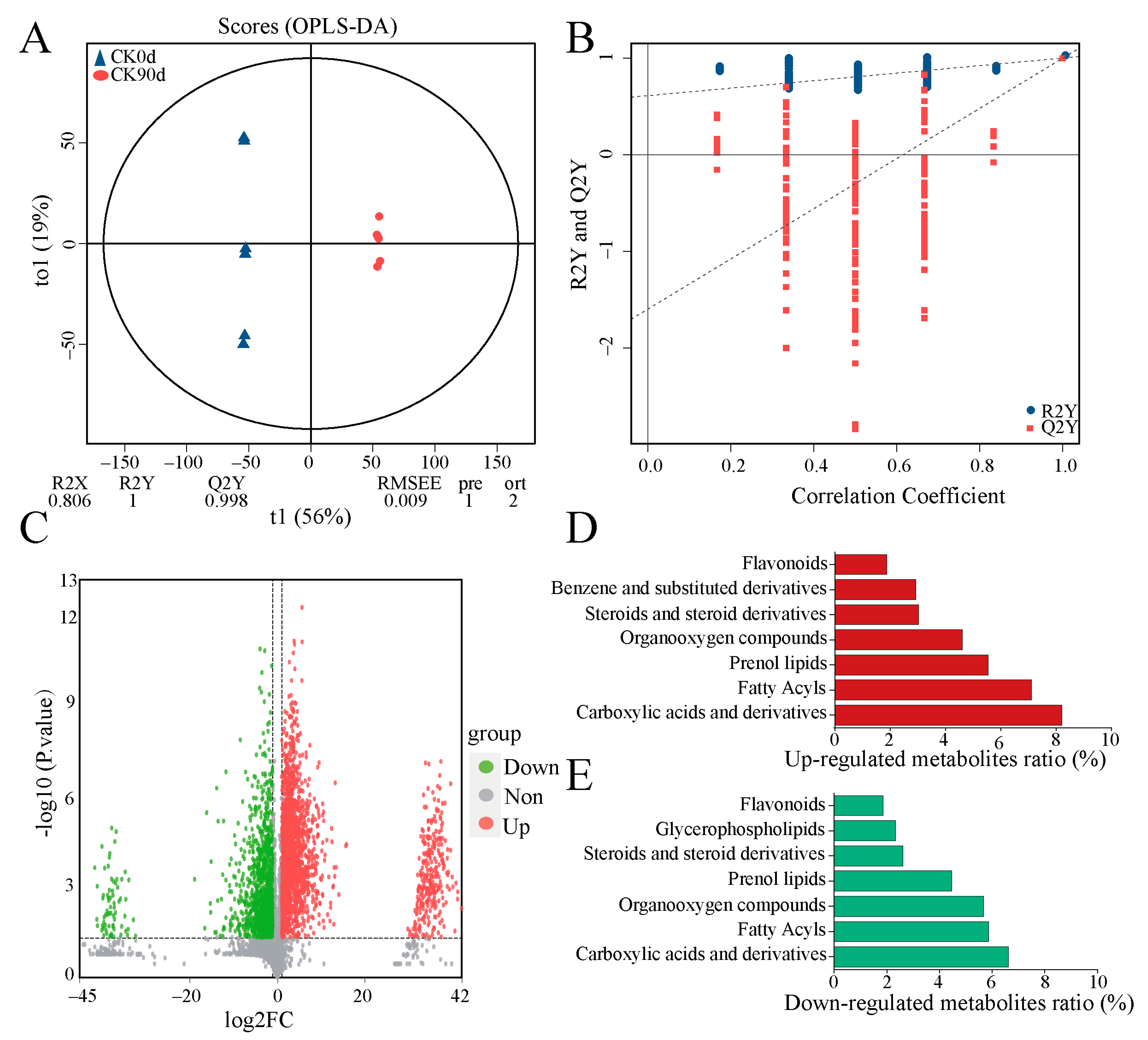
3.5. Metabolite Composition of Mixed Silage at 0 Days and 90 Days
3.6. Combined Analysis of Microorganisms and Metabolites
4. Discussion
4.1. Fermentation Characteristics and Chemical Content of Mixed Feeds of Oats and Vetch
4.2. Bacterial Composition of Mixed Oats and Vetch
4.3. Fungal Composition of Mixed Oats and Vetch
4.4. Metabolite Composition of Mixed Oats and Vetch
4.5. Microbial and Metabolite Interactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cai, L.; Zhao, J.; Chen, J. Coevolutionary dynamics in the grass-livestock social-ecological system of China’s alpine pastoral areas: A case study of the Qilian Mountains region in China. PLoS ONE 2025, 20, e0317769. [Google Scholar] [CrossRef]
- Yu, H.; Yang, Q.; Jiang, S.; Zhan, B.; Zhan, C. Detection and Attribution of Vegetation Dynamics in the Yellow River Basin Based on Long-Term Kernel NDVI Data. Remote Sens. 2024, 16, 1280. [Google Scholar] [CrossRef]
- Gustine, R.; Theophilus, A.; Flint, C.G.; Ulrich-Schad, J.D.; Epperson, E.; Wright, C.M.; Adam, J.C. Managing beyond water: Utilizing community well-being interviews in the Upper Yakima River Basin, USA, for climate change adaptation. Ecol. Soc. 2025, 30, 7. [Google Scholar] [CrossRef]
- Ma, T.; Xin, Y.; Chen, X.; Wen, X.; Wang, F.; Liu, H.; Zhu, L.; Li, X.; You, M.; Yan, Y. Effects of Compound Lactic Acid Bacteria Additives on the Quality of Oat and Common Vetch Silage in the Northwest Sichuan Plateau. Fermentation 2025, 11, 93. [Google Scholar] [CrossRef]
- Ji, J.; Wang, Z.; Gao, P.; Tan, X.; Wang, X.; Kuai, J.; Wang, J.; Xu, Z.; Wang, B.; Zhou, G.; et al. Rapeseed Supports Hairy Vetch in Intercropping, Enhancing Root and Stem Morphology, Nitrogen Metabolism, Photosynthesis, and Forage Yield. Agronomy 2025, 15, 220. [Google Scholar] [CrossRef]
- Rady, A.M.; Attia, M.F.; Kholif, A.E.; Sallam, S.M.; Vargas-Bello-Pérez, E. Improving fodder yields and nutritive value of some forage grasses as animal feeds through intercropping with Egyptian clover (Trifolium alexandrinum L.). Agronomy 2022, 12, 2589. [Google Scholar] [CrossRef]
- Du, E.; Mao, N.; Liu, S.; Zhang, H.; Fan, M.; Sun, H.; Zheng, Y.; Cheng, Q.; Wang, C.; Li, P.; et al. Effects of different wet distillers’ grains ratios on fermentation quality, nitrogen fractions and bacterial communities of total mixed ration silage. BMC Microbiol. 2025, 25, 31. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hao, J.; Zhao, M.; Yan, X.; Jia, Y.; Wang, Z.; Ge, G. Effects of different temperature and density on quality and microbial population of wilted alfalfa silage. BMC Microbiol. 2024, 24, 380. [Google Scholar] [CrossRef]
- Anumudu, C.K.; Miri, T.; Onyeaka, H. Multifunctional Applications of Lactic Acid Bacteria: Enhancing Safety, Quality, and Nutritional Value in Foods and Fermented Beverages. Foods 2024, 13, 3714. [Google Scholar] [CrossRef]
- Storm, I.M.D.; Rasmussen, R.R.; Rasmussen, P.H. Occurrence of pre-and post-harvest mycotoxins and other secondary metabolites in Danish maize silage. Toxins 2014, 6, 2256–2269. [Google Scholar] [CrossRef]
- Wu, B.; Ren, T.; Li, C.; Wu, S.; Cao, X.; Mei, H.; Wu, T.; Yong, M.; Wei, M.; Wang, C. Exploring the Fermentation Products, Microbiology Communities, and Metabolites of Big-Bale Alfalfa Silage Prepared with/without Molasses and Lactobacillus rhamnosus. Agriculture 2024, 14, 1560. [Google Scholar] [CrossRef]
- Kozloski, G.V.; Senger, C.C.D.; Perottoni, J.; Sanchez, L.B. Evaluation of two methods for ammonia extraction and analysis in silage samples. Anim. Feed Sci. Technol. 2006, 127, 336–342. [Google Scholar] [CrossRef]
- Enne, G.; Elez, D.; Fondrini, F.; Bonizzi, I.; Feligini, M.; Aleandri, R. High-performance liquid chromatography of governing liquid to detect illegal bovine milk’s addition in water buffalo mozzarella: Comparison with results from raw milk and cheese matrix. J. Chromatogr. A 2005, 1094, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Association of Official Analytical Chemists. Official Methods of Analysis of the Association of Official Analytical Chemists; The Association of Official Analytical Chemists: Gaithersburg, MD, USA, 2000; Volume 11. [Google Scholar]
- Van Soest, P.V.; Robertson, J.B.; Lewis, B.A. Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef]
- (ASTM) D 1102–84; Standard Test Method for Ash in Wood. American Society for Testing and Materials, ASTM International: West Conshohocken, PA, USA, 2021. [CrossRef]
- Liu, C.; Zhao, D.; Ma, W.; Guo, Y.; Wang, A.; Wang, Q.; Lee, D.J. Denitrifying sulfide removal process on high-salinity wastewaters in the presence of Halomonas sp. Appl. Microbiol. Biotechnol. 2016, 100, 1421–1426. [Google Scholar] [CrossRef]
- Scibetta, S.; Schena, L.; Abdelfattah, A.; Pangallo, S.; Cacciola, S.O. Selection and experimental evaluation of universal primers to study the fungal microbiome of higher plants. Phytobiomes J. 2018, 2, 225–236. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; DeBoy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef]
- Chen, H.; Jiang, W. Application of high-throughput sequencing in understanding human oral microbiome related with health and disease. Front. Microbiol. 2014, 5, 508. [Google Scholar] [CrossRef]
- Li, R.; Sun, Z.; Zhao, Y.; Li, L.; Yang, X.; Cen, J.; Chen, S.; Li, C.; Wang, Y. Application of UHPLC-Q-TOF-MS/MS metabolomics approach to investigate the taste and nutrition changes in tilapia fillets treated with different thermal processing methods. Food Chem. 2021, 356, 129737. [Google Scholar] [CrossRef]
- Guan, H.; Shuai, Y.; Ran, Q.; Yan, Y.; Wang, X.; Li, D.; Cai, Y.; Zhang, X. The microbiome and metabolome of Napier grass silages prepared with screened lactic acid bacteria during ensiling and aerobic exposure. Anim. Feed Sci. Technol. 2020, 269, 114673. [Google Scholar] [CrossRef]
- He, C.; Li, Q.; Xiao, H.; Sun, X.; Gao, Z.; Cai, Y.; Zhao, S. Effects of mixing ratio and lactic acid bacteria preparation on the quality of whole-plant quinoa and whole-plant corn or stevia powder mixed silage. Microorganisms 2025, 13, 78. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Koli, P.; Singh, T.; Das, M.M.; Maity, S.B.; Singh, K.K.; Katiyar, R.; Misra, A.K.; Mahanta, S.K.; Srivastava, M.K.; et al. Assessing genotypes of buffel grass (cenchrus ciliaris) as an alternative to maize silage for sheep nutrition. PLoS ONE 2024, 19, e0304328. [Google Scholar] [CrossRef]
- Souza, M.S.; de Queiroz, A.C.M.; Bernardes, T.F.; Faturi, C.; Domingues, F.N.; Rodrigues, J.P.P.; da Silva, T.C.; Rêgo, A.C.D. Effects of sodium benzoate application, silage relocation, and storage time on the preservation quality of sugarcane silage. Agronomy 2022, 12, 1533. [Google Scholar] [CrossRef]
- Chen, F.; Wang, J.; Zhang, S.; Abdul, S.C.; Khanaki, H. Assessing fermentation quality, aerobic stability, in vitro digestibility, and rumen degradation characteristics of silages mixed with sweet sorghum and aerial parts of licorice. Agriculture 2024, 14, 212. [Google Scholar] [CrossRef]
- Zheng, M.; Li, Q.; Mao, P.; Tian, X.; Guo, Y.; Lin, M. Identification and correlation analysis of key clostridia and LAB species in alfalfa silages prepared with different cultivars and additives. Agriculture 2024, 14, 1963. [Google Scholar] [CrossRef]
- Serva, L. A comparative evaluation of maize silage quality under diverse pre-ensiling strategies. PLoS ONE 2024, 19, e0308627. [Google Scholar] [CrossRef]
- Blajman, J.E.; Vinderola, G.; Páez, R.B.; Signorini, M.L. The role of homofermentative and heterofermentative lactic acid bacteria for alfalfa silage: A meta-analysis. J. Agric. Sci. 2020, 158, 107–118. [Google Scholar] [CrossRef]
- Murniece, R.; Reidzane, S.; Radenkovs, V.; Straumite, E.; Keke, A.; Kobrin, E.; Klava, D. Scald fermentation time as a factor determining the nutritional and sensory quality of rye bread. Foods 2025, 14, 979. [Google Scholar] [CrossRef]
- Xin, Y.; Chen, C.; Zhong, Y.; Bu, X.; Huang, S.; Tahir, M.; Du, Z.; Liu, W.; Yang, W.; Li, J.; et al. Effect of storage time on the silage quality and microbial community of mixed maize and faba bean in the Qinghai-Tibet Plateau. Front. Microbiol. 2023, 13, 1090401. [Google Scholar] [CrossRef]
- Mao, K.; Yu, Z.; Huang, S.; Wang, M.; Hannaway, D.B. Effect of storage period on the fermentation profile and bacterial community of silage prepared with alfalfa, whole-plant corn and their mixture. Fermentation 2022, 8, 486. [Google Scholar] [CrossRef]
- Wang, X.; Song, J.; Liu, Z.; Zhang, G.; Zhang, Y. Fermentation quality and microbial community of corn stover or rice straw silage mixed with soybean curd residue. Animals 2022, 12, 919. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Drouin, P.; Lafrenière, C. Effect of temperature (5–25 °C) on epiphytic lactic acid bacteria populations and fermentation of whole-plant corn silage. J. Appl. Microbiol. 2016, 121, 657–671. [Google Scholar] [CrossRef]
- Bak, K.H.; Bauer, S.; Eisenreich, C.; Paulsen, P. Residual Nitrite, Nitrate, and Volatile N-Nitrosamines in Organic and Conventional Ham and Salami Products. Foods 2025, 14, 112. [Google Scholar] [CrossRef]
- Hord, N.G.; Tang, Y.; Bryan, N.S. Food sources of nitrates and nitrites: The physiologic context for potential health benefits. Am. J. Clin. Nutr. 2009, 90, 1–10. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, L.; Zhang, B.; Bao, J. pH shifting adaptive evolution stimulates the low pH tolerance of Pediococcus acidilactici and high L-lactic acid fermentation efficiency. Bioresour. Technol. 2025, 416, 131813. [Google Scholar] [CrossRef]
- Meng, H.; Jiang, Y.; Wang, L.; Li, Y.; Wang, S.; Tong, X.; Wang, S. Dynamic Analysis of Fermentation Quality, Microbial Community, and Metabolome in the Whole Plant Soybean Silage. Fermentation 2024, 10, 535. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Yang, X.; Huang, L.; Wang, P.; Zhao, L. Effects of Lactiplantibacillus plantarum and Fermentation Time on the Quality, Bacterial Community, and Functional Prediction of Silage from Lotus corniculatus L. in Karst Regions. Agriculture 2024, 15, 16. [Google Scholar] [CrossRef]
- Yin, H.; Zhao, M.; Yang, R.; Sun, J.; Yu, Z.; Bai, C.; Xue, Y. Effect of Regulation of Whole-Plant Corn Silage Inoculated with Lactobacillus buchneri or Bacillus licheniformis Regarding the Dynamics of Bacterial and Fungal Communities on Aerobic Stability. Plants 2024, 13, 1471. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, G.; Wang, L.; Zhang, B.; Chen, J.; Liu, Y.; Pang, H.; Tan, Z. Characteristics of Lactobacillus plantarum QZW5 and its effects on wheat silage under multigelation. Chem. Biol. Technol. Agric. 2021, 8, 52. [Google Scholar] [CrossRef]
- Li, Y.; Da Silva, E.B.; Li, J.; Kung, L., Jr. Effect of homo-fermentative lactic acid bacteria inoculants on fermentation characteristics and bacterial and fungal communities in alfalfa silage. Fermentation 2022, 8, 621. [Google Scholar] [CrossRef]
- Kong, F.; Lu, N.; Liu, Y.; Zhang, S.; Jiang, H.; Wang, H.; Wang, W.; Li, S. Aspergillus oryzae and Aspergillus niger co-cultivation extract affects in vitro degradation, fermentation characteristics, and bacterial composition in a diet-specific manner. Animals 2021, 11, 1248. [Google Scholar] [CrossRef] [PubMed]
- Sha, T.; Lu, Y.; He, P.; Hassan, M.M.; Tong, Y. Recent Advances in Physicochemical Control and Potential Green Ecologic Strategies Related to the Management of Mold in Stored Grains. Foods 2025, 14, 961. [Google Scholar] [CrossRef]
- Castañeda-Casasola, C.C.; Nieto-Jacobo, M.F.; Soares, A.; Padilla-Padilla, E.A.; Anducho-Reyes, M.A.; Brown, C.; Soth, S.; Esquivel-Naranjo, E.U.; Hampton, J.; Mendoza-Mendoza, A. Unveiling a Microexon Switch: Novel Regulation of the Activities of Sugar Assimilation and Plant-Cell-Wall-Degrading Xylanases and Cellulases by Xlr2 in Trichoderma virens. Int. J. Mol. Sci. 2024, 25, 5172. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tan, Z.; Gu, L.; Ma, H.; Wang, Z.; Wang, L.; Wu, G.; Qin, G.; Wang, Y.; Pang, H. Variation of microbial community and fermentation quality in corn silage treated with lactic acid bacteria and Artemisia argyi during aerobic exposure. Toxins 2022, 14, 349. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Zhou, P.; Yue, Z.; Wang, Z.; Qin, G.; Wang, Y.; Tan, Z.; Cai, Y. Fermentation characteristics, chemical composition, and aerobic stability in whole crop corn silage treated with lactic acid bacteria or Artemisia argyi. Agriculture 2024, 14, 1015. [Google Scholar] [CrossRef]
- Gu, Q.; Zhang, J.; Lin, B.; Ding, H.; Yan, Q.; Wei, C.; Yao, Y.; Wang, R.; Zou, C. Effects and function of citric acid on fermentation quality and microbial community in sugarcane tops silage with high and low water-soluble carbohydrate content. BMC Plant Biol. 2025, 25, 99. [Google Scholar] [CrossRef]
- Liu, B.; Yang, Z.; Huan, H.; Gu, H.; Xu, N.; Ding, C. Impact of molasses and microbial inoculants on fermentation quality, aerobic stability, and bacterial and fungal microbiomes of barley silage. Sci. Rep. 2020, 10, 5342. [Google Scholar] [CrossRef]
- Liu, Y.; Bao, J.; Si, Q.; Liu, M.; Bai, B.; Fu, Z.; Ge, G.; Jia, Y.; Wang, Z. Effects of lactic acid bacteria additives on fatty acids, amino acids and antioxidant capacity of Leymus chinensis silage during aerobic exposure. Fermentation 2023, 9, 323. [Google Scholar] [CrossRef]
- Zhang, Z.; Han, H.; Zhao, J.; Liu, Z.; Deng, L.; Wu, L.; Niu, J.; Guo, Y.; Wang, G.; Gou, X.; et al. Peptide hormones in plants. Mol. Hortic. 2025, 5, 7. [Google Scholar] [CrossRef]
- Hao, Y.; Jiang, X.; Sun, R.; Bai, Y.; Xu, C.; Song, Y.; Xia, C. Effects of Supplementing Rumen-Protected Glutathione on Lactation Performance, Nutrients, Oxidative Stress, Inflammation, and Health in Dairy Cows During the Transition Period. Vet. Sci. 2025, 12, 84. [Google Scholar] [CrossRef]
- Mansour, M.A.; Ali, S.G.; Hassan, M.A.; Gabra, F.A.; Mawad, A.M. Optimization of citrulline production from a Bacillus subtilis BH-01 isolated from raw buffalo milk. BMC Microbiol. 2025, 25, 71. [Google Scholar] [CrossRef] [PubMed]
- Chamberlin, A.; Mitsuhashi, Y.; Bigley, K.; Bauer, J.E. Unexpected depletion of plasma arachidonate and total protein in cats fed a low arachidonic acid diet due to peroxidation. Br. J. Nutr. 2011, 106, S131–S134. [Google Scholar] [CrossRef]
- Mititelu, M.; Lupuliasa, D.; Neacșu, S.M.; Olteanu, G.; Busnatu, Ș.S.; Mihai, A.; Popovici, V.; Măru, N.; Boroghină, S.C.; Mihai, S.; et al. Polyunsaturated Fatty Acids and Human Health: A Key to Modern Nutritional Balance in Association with Polyphenolic Compounds from Food Sources. Foods 2024, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Hastings, N.; Agaba, M.K.; Tocher, D.R.; Zheng, X.; Dickson, C.A.; Dick, J.R.; Teale, A.J. Molecular cloning and functional characterization of fatty acyl desaturase and elongase cDNAs involved in the production of eicosapentaenoic and docosahexaenoic acids from α-linolenic acid in Atlantic salmon (Salmo salar). Mar. Biotechnol. 2004, 6, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wu, N.; Na, N.; Ding, H.; Sun, L.; Fang, Y.; Li, D.; Li, E.; Yang, B.; Wei, X.; et al. Dynamics of fermentation quality, bacterial communities, and fermentation weight loss during fermentation of sweet sorghum silage. BMC Microbiol. 2024, 24, 429. [Google Scholar] [CrossRef]
- Qin, X.; Fan, Z.; Qiao, S.; Li, J.; Lv, J. Genetically predicted plasma metabolites mediate the causal relationship between gut microbiota and osteosarcoma. Sci. Rep. 2025, 15, 7277. [Google Scholar] [CrossRef]
- Su, H.; Wang, Z.; Ma, L.; Qin, R.; Chang, T.; Zhang, Z.; Yao, J.; Li, X.; Li, S.; Hu, X.; et al. Multitrophic Diversity of the Biotic Community Drives Ecosystem Multifunctionality in Alpine Grasslands. Ecol. Evol. 2024, 14, e70511. [Google Scholar] [CrossRef]
- Holtshausen, L.; Beauchemin, K.A. Supplementing barley-based dairy cow diets with Saccharomyces cerevisiae. Prof. Anim. Sci. 2010, 26, 285–289. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Content |
|---|---|
| pH | 4.12 |
| AN (%TN) | 16.86 |
| LA (%DM) | 1.12 |
| AA (%DM) | 0.22 |
| PA (%DM) | 0.00 |
| Treatment | Items | ||||||
|---|---|---|---|---|---|---|---|
| DM (%) | CP (% DM) | WSC (% DM) | NDF (% DM) | ADF (% DM) | Ash (% DM) | EE (% DM) | |
| CK0d | 37.74 ± 0.26 a | 13.85 ± 0.13 a | 13.00 ± 0.76 a | 50.09 ± 0.46 a | 30.40 ± 0.19 a | 6.72 ± 0.12 b | 2.76 ± 0.03 b |
| CK90d | 34.41 ± 0.11 b | 10.58 ± 0.10 b | 8.77 ± 0.03 b | 45.08 ± 0.09 b | 24.97 ± 0.10 b | 11.05 ± 0.11 a | 3.57 ± 0.17 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Yin, G.; Yu, X. Dynamics of Microorganisms and Metabolites in the Mixed Silage of Oats and Vetch in Alpine Pastures, and Their Regulatory Mechanisms Under Low Temperatures. Microorganisms 2025, 13, 1535. https://doi.org/10.3390/microorganisms13071535
Xu S, Yin G, Yu X. Dynamics of Microorganisms and Metabolites in the Mixed Silage of Oats and Vetch in Alpine Pastures, and Their Regulatory Mechanisms Under Low Temperatures. Microorganisms. 2025; 13(7):1535. https://doi.org/10.3390/microorganisms13071535
Chicago/Turabian StyleXu, Shuangpeng, Guoli Yin, and Xiaojun Yu. 2025. "Dynamics of Microorganisms and Metabolites in the Mixed Silage of Oats and Vetch in Alpine Pastures, and Their Regulatory Mechanisms Under Low Temperatures" Microorganisms 13, no. 7: 1535. https://doi.org/10.3390/microorganisms13071535
APA StyleXu, S., Yin, G., & Yu, X. (2025). Dynamics of Microorganisms and Metabolites in the Mixed Silage of Oats and Vetch in Alpine Pastures, and Their Regulatory Mechanisms Under Low Temperatures. Microorganisms, 13(7), 1535. https://doi.org/10.3390/microorganisms13071535