
FpFumB Is Required for Basic Biological Processes and Virulence in Fusarium proliferatum by Modulating DNA Repair Through Interaction with FpSae2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Strains and Culture Conditions
2.2. Gene Deletion and Complementation Constructs
2.3. Virulence Assays in Medicago Sativa
2.4. RNA Extraction and Quantitative PCR
2.5. RNA Sequencing
2.6. Western Blot
2.7. Yeast Two-Hybrid Assay
2.8. Co-Immunoprecipitation Assay
2.9. Mitochondrial Function and Metabolic Assays
2.10. Protein Prokaryotic Expression
3. Results
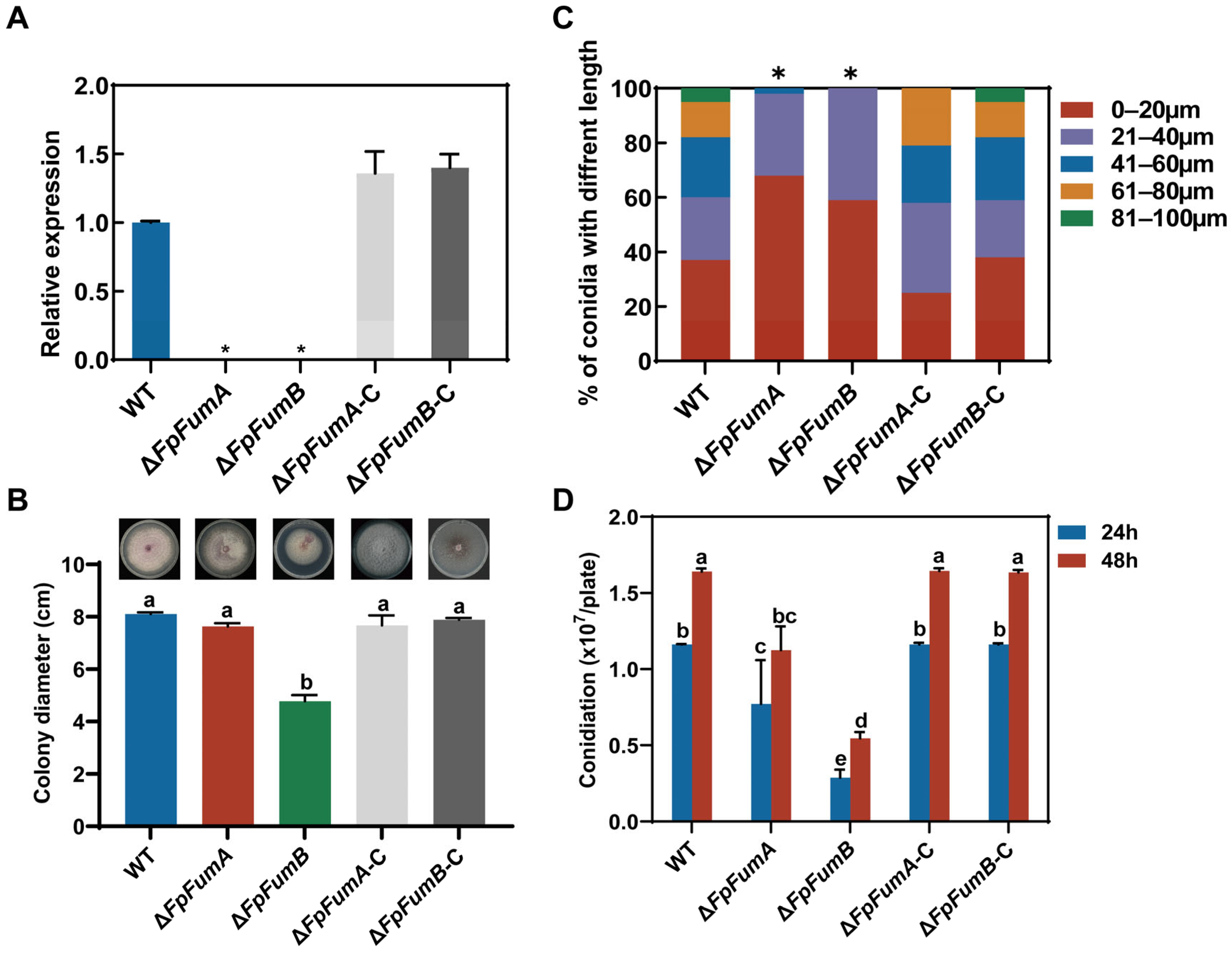
3.1. Regulation of FpFUM Genes on Basic Biological Processes of Fusarium proliferatum
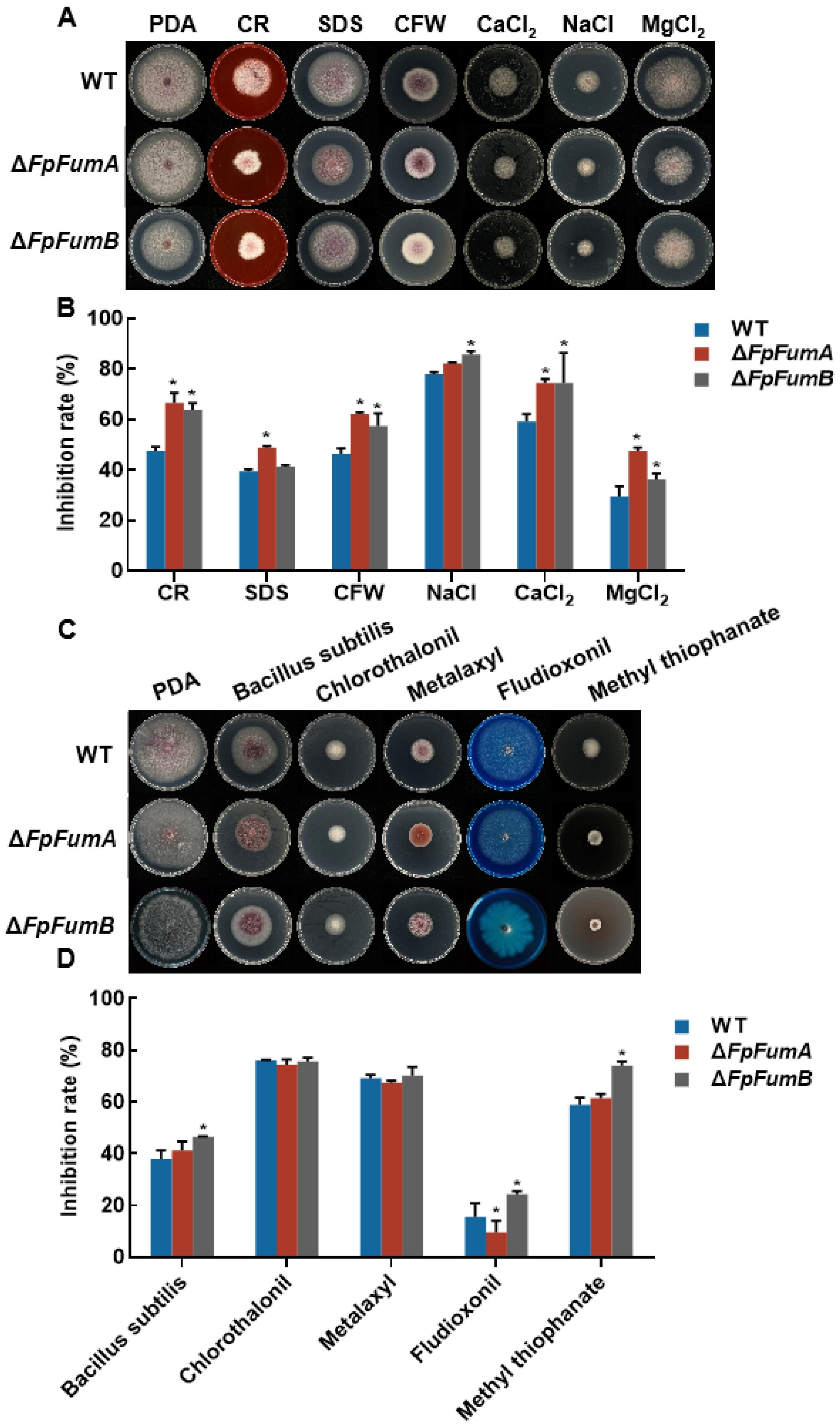
3.2. FpFUM Impacts Environmental Stress Response in Fusarium proliferatum
3.3. Role of FpFumB in Regulating Pathogenicity of Fusarium proliferatum

3.4. Impact of FpFUM Gene on Mitochondrial Function and Metabolism in Fusarium proliferatum
3.5. Exogenous Malate Restores the Phenotypic Defect in ΔFpFumB Mutant
3.6. Identification of Key Amino Acid Sites in FpFumB Protein
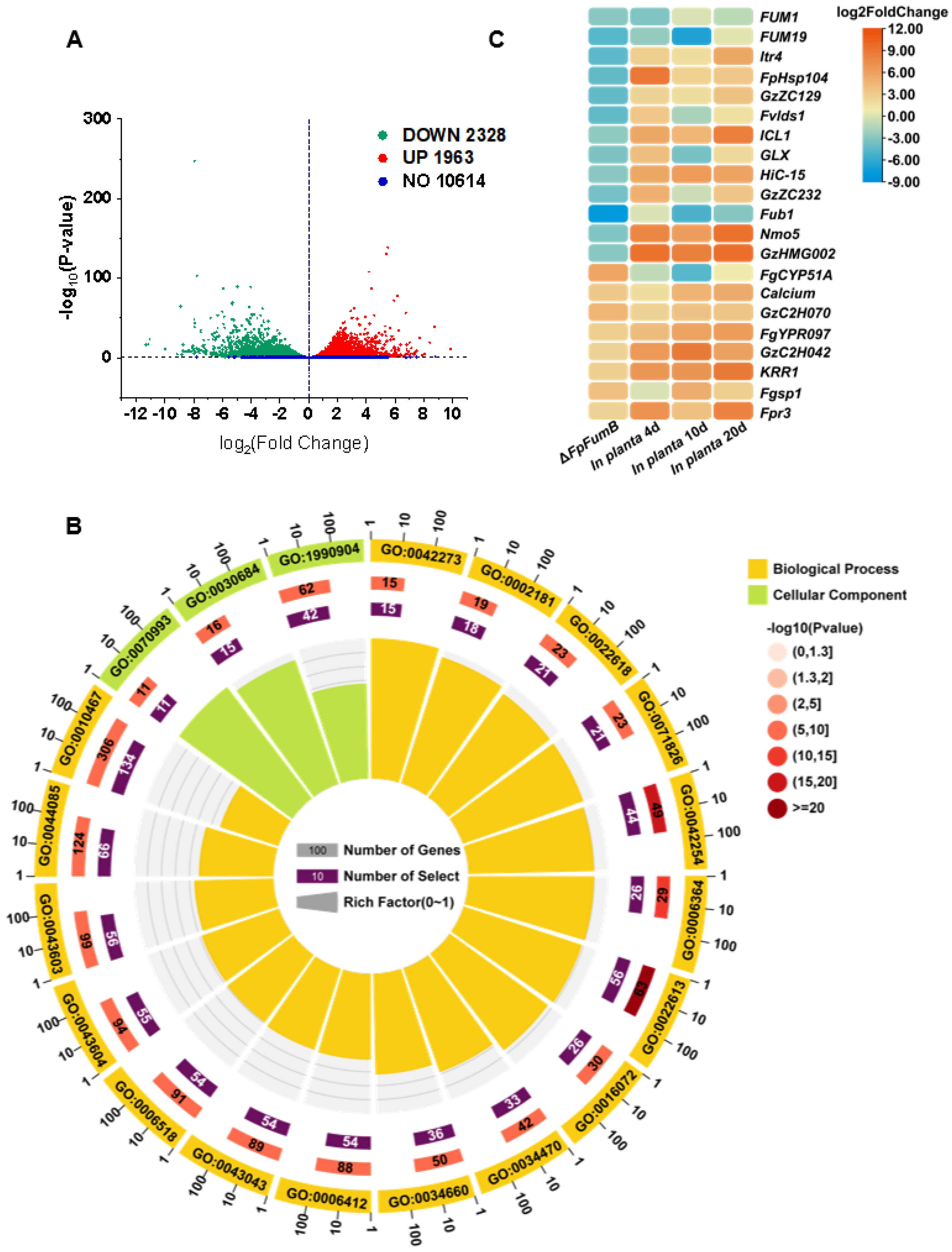
3.7. Functional Characterization of Gene Groups Regulated by the FpFumB Gene in Fusarium proliferatum
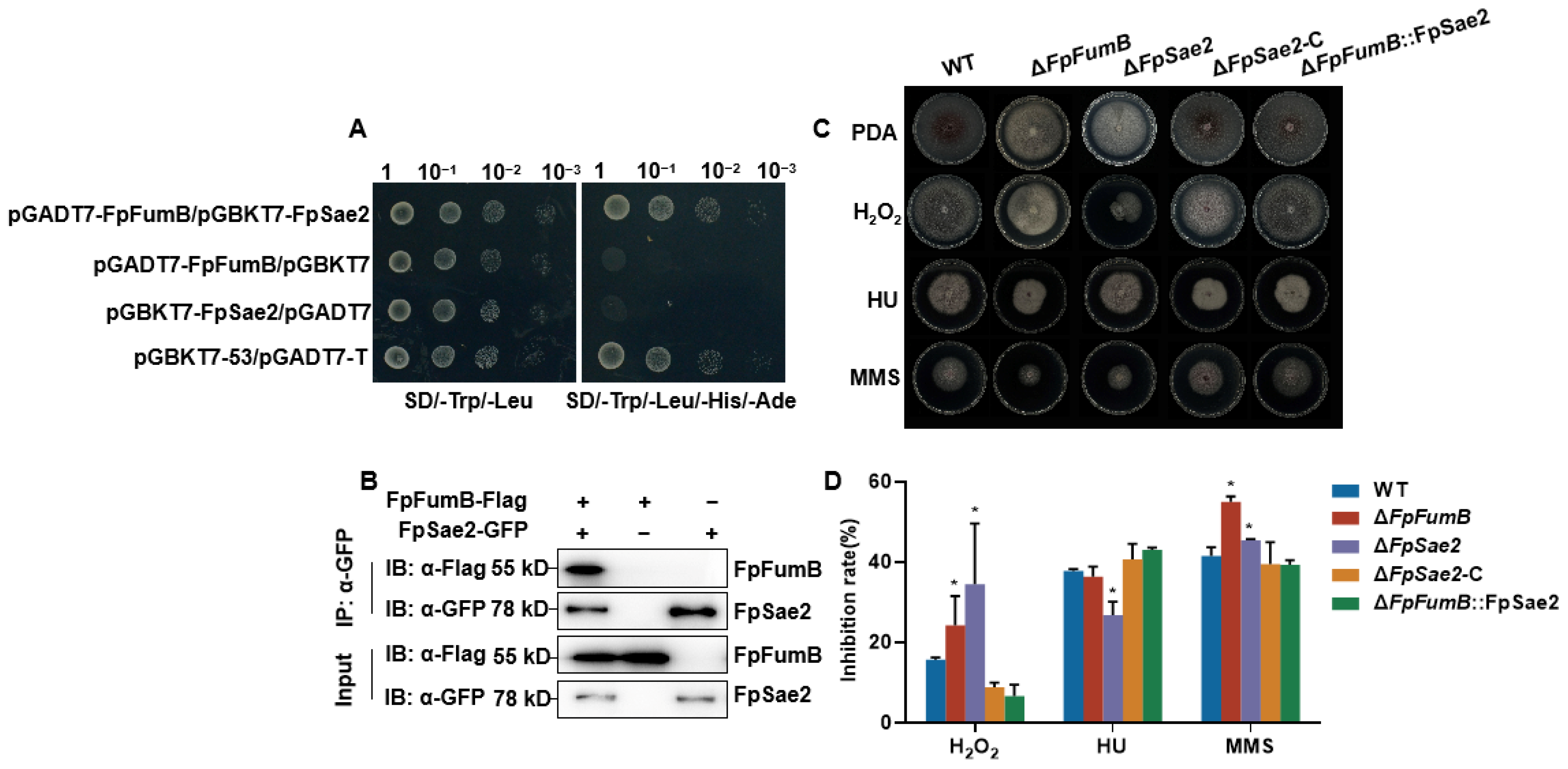
3.8. FpFumB Regulates DNA Damage Repair Function
3.9. Role of FpSae2 in Regulating Growth, Sporulation, and Pathogenicity of Fusarium proliferatum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CWDEs | cell-wall-degrading enzymes |
| ROS | reactive oxygen species |
| VOCs | volatile organic compounds |
| TCA | tricarboxylic acid |
| WT | wild-type |
| PDA | potato dextrose agar |
| PDB | potato dextrose broth |
| CMC | carboxymethyl cellulose |
| CR | Congo Red |
| CFW | Calcofluor White |
| SDS | sodium dodecyl sulfate |
| HU | hydroxyurea |
| MMS | methyl methanesulfonate |
| qPCR | quantitative PCR |
| WA | water agar |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| RLU | relative light units |
| IPTG | isopropyl β-D-1-thiogalactopyranoside |
| DEGs | differentially expressed genes |
| H2O2 | hydrogen peroxide |
References
- Ajmal, M.; Hussain, A.; Ali, A.; Chen, H.; Lin, H. Strategies for Controlling the Sporulation in Fusarium spp. J. Fungi 2022, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Pérez, B.A.; Berretta, M.F.; Carrión, E.; Wright, E.R. First report of root rot caused by Fusarium proliferatum on blueberry in argentina. Plant Dis. 2011, 95, 1478. [Google Scholar] [CrossRef] [PubMed]
- Munkvold, G.P.; Proctor, R.H.; Moretti, A. Mycotoxin production in Fusarium according to contemporary species concepts. Annu. Rev. Phytopathol. 2021, 59, 373–402. [Google Scholar] [CrossRef] [PubMed]
- Kamle, M.; Mahato, D.K.; Devi, S.; Lee, K.E.; Kang, S.G.; Kumar, P. Fumonisins: Impact on agriculture, food, and human health and their management strategies. Toxins 2019, 11, 328. [Google Scholar] [CrossRef]
- Wang, A.; Haapalainen, M.; Latvala, S.; Edelenbos, M.; Johansen, A. Discriminant analysis of volatile organic compounds of Fusarium oxysporum f. sp. cepae and Fusarium proliferatum isolates from onions as indicators of fungal growth. Fungal Biol. 2018, 122, 1013–1022. [Google Scholar] [CrossRef]
- Sharafaddin, A.H.; Hamad, Y.K.; El Komy, M.H.; Ibrahim, Y.E.; Widyawan, A.; Molan, Y.Y.; Saleh, A.A. Cell wall degrading enzymes and their impact on Fusarium proliferatum pathogenicity. Eur. J. Plant Pathol. 2019, 155, 871–880. [Google Scholar] [CrossRef]
- Dobosz, B.; Drzewiecka, K.; Waskiewicz, A.; Irzykowska, L.; Bocianowski, J.; Karolewski, Z.; Kostecki, M.; Kruczynski, Z.; Krzyminiewski, R.; Weber, Z.; et al. Free Radicals, salicylic acid and mycotoxins in Asparagus after inoculation with Fusarium proliferatum and F. oxysporum. Appl. Magn. Reson. 2011, 41, 19–30. [Google Scholar] [CrossRef]
- Xie, L.; Yang, Q.; Wu, Y.; Xiao, J.; Qu, H.; Jiang, Y.; Li, T. Fumonisin B1 biosynthesis is associated with oxidative stress and plays an important role in Fusarium proliferatum infection on banana fruit. J. Agric. Food Chem. 2023, 71, 5372–5381. [Google Scholar] [CrossRef]
- Fanelli, F.; Schmidt-Heydt, M.; Haidukowski, M.; Geisen, R.; Logrieco, A.; Mulè, G. Influence of light on growth, fumonisin biosynthesis and FUM1 gene expression by Fusarium proliferatum. Int. J. Food Microbiol. 2012, 153, 148–153. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, Y.; He, S.; Li, H.; Wang, Y.; Wu, Z. FpOGT is required for fungal growth, stress response, and virulence of Fusarium proliferatum by affecting the expression of glucokinase and other glucose metabolism-related genes. Phytopathol. Res. 2024, 6, 2. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, Q.; Xie, L.; Yin, C.; Qu, H.; He, J.; Jiang, Y.; Li, T. Possible mechanism of contribution of a secreted aspartic proteinase FpOPSB to the virulence of Fusarium proliferatum causing banana crown rot. Food Front. 2023, 4, 1511–1522. [Google Scholar] [CrossRef]
- Zhao, P.B.; Ren, A.Z.; Li, D.C. The FUS3/KSS1-type MAP kinase gene FPK1 is involved in hyphal growth, conidiation and plant infection of Fusarium proliferatum. J. Mol. Microbiol. Biotechnol. 2011, 21, 110–119. [Google Scholar]
- Leshets, M.; Silas, Y.; Lehming, N.; Pines, O. Fumarase: From the TCA Cycle to DNA damage response and tumor suppression. Front. Mol. Biosci. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Kavanagh, K.L.; Yue, W.W.; Lee, W.H.; Muller-Knapp, S.; Gileadi, O.; Sacchettini, J.; Oppermann, U. Structural basis of fumarate hydratase deficiency. J. Inherit. Metab. Dis. 2011, 34, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Todisco, S.; Convertini, P.; Iacobazzi, V.; Infantino, V. TCA Cycle rewiring as emerging metabolic Signature of hsepatocellular carcinoma. Cancers 2020, 12, 68. [Google Scholar]
- Sun, J.K.; Wong, G.C.; Chow, K.H. Cross-talk between DNA damage response and the central carbon metabolic network underlies selective vulnerability of Purkinje neurons in ataxia-telangiectasia. J. Neurochem. 2023, 166, 654–677. [Google Scholar] [CrossRef]
- Sobanski, T.; Rose, M.; Suraweera, A.; O’Byrne, K.; Richard, D.J.; Bolderson, E. Cell metabolism and DNA repair pathways: Implications for cancer therapy. Front. Cell Dev. Biol. 2021, 9, 633305. [Google Scholar] [CrossRef]
- Pracharoenwattana, I.; Zhou, W.; Keech, O.; Francisco, P.B.; Udomchalothorn, T.; Tschoep, H.; Stitt, M.; Gibon, Y.; Smith, S.M. Arabidopsis has a cytosolic fumarase required for the massive allocation of photosynthate into fumaric acid and for rapid plant growth on high nitrogen. Plant J. 2010, 62, 785–795. [Google Scholar] [CrossRef]
- Saunders, H.A.; Calzadilla, P.I.; Schwartz, J.M.; Johnson, G.N. Cytosolic fumarase acts as a metabolic fail-safe for both high and low temperature acclimation of Arabidopsis thaliana. J. Exp. Bot. 2022, 73, 2112–2124. [Google Scholar] [CrossRef]
- Zubimendi, J.P.; Martinatto, A.; Valacco, M.P.; Moreno, S.; Andreo, C.S.; Drincovich, M.F.; Tronconi, M.A. The complex allosteric and redox regulation of the fumarate hydratase and malate dehydratase reactions of Arabidopsis thaliana Fumarase 1 and 2 gives clues for understanding the massive accumulation of fumarate. FEBS J. 2018, 285, 2205–2224. [Google Scholar] [CrossRef]
- van der Merwe, M.J.; Osorio, S.; Moritz, T.; Nunes-Nesi, A.; Fernie, A.R. Decreased mitochondrial activities of malate dehydrogenase and fumarase in tomato lead to altered root growth and architecture via diverse mechanisms. Plant Physiol. 2009, 149, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Silas, Y.; Singer, E.; Das, K.; Lehming, N.; Pines, O. A combination of Class-I fumarases and metabolites (α-ketoglutarate and fumarate) signal the DNA damage response in Escherichia coli. Proc. Natl. Acad. Sci. USA 2021, 118, e2026595118. [Google Scholar] [CrossRef] [PubMed]
- Hassett, D.J.; Howell, M.L.; Ochsner, U.A.; Vasil, M.L.; Johnson, Z.; Dean, G.E. An operon containing fumC and sodA encoding fumarase C and manganese superoxide dismutase is controlled by the ferric uptake regulator in Pseudomonas aeruginosa: Fur mutants produce elevated alginate levels. J. Bacteriol. 1997, 179, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Yip, J.; Wang, S.; Tan, J.; Lim, T.; Lin, Q.; Karmon, O.; Pines, O.; Lehming, N. Fumarase affects the DNA damage response by protecting the mitochondrial desulfurase Nfs1p from modification and inactivation. iScience 2021, 24, 103354. [Google Scholar] [CrossRef]
- Leshets, M.; Ramamurthy, D.; Lisby, M.; Lehming, N.; Pines, O. Fumarase is involved in DNA double-strand break resection through a functional interaction with Sae2. Curr. Genet. 2018, 64, 697–712. [Google Scholar] [CrossRef]
- Clerici, M.; Mantiero, D.; Lucchini, G.; Longhese, M.P. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J. Biol. Chem. 2005, 280, 38631–38638. [Google Scholar] [CrossRef]
- Giallongo, S.; Costa, F.; Longhitano, L.; Giallongo, C.; Ferrigno, J.; Tropea, E.; Vicario, N.; Li Volti, G.; Parenti, R.; Barbagallo, I.; et al. The pleiotropic effects of fumarate: From mitochondrial respiration to epigenetic rewiring and DNA repair mechanisms. Metabolites 2023, 13, 880. [Google Scholar] [CrossRef]
- Yogev, O.; Yogev, O.; Singer, E.; Shaulian, E.; Goldberg, M.; Fox, T.D.; Pines, O. Fumarase: A mitochondrial metabolic enzyme and a cytosolic/nuclear component of the DNA damage response. PLoS Biol. 2010, 8, e1000328. [Google Scholar] [CrossRef]
- Justino, M.C.; Almeida, C.C.; Teixeira, M.; Saraiva, L.M. Escherichia coli di-iron YtfE protein is necessary for the repair of stress-damaged iron-sulfur clusters. J. Biol. Chem. 2007, 282, 10352–10359. [Google Scholar] [CrossRef]
- Silas, Y.; Singer, E.; Lehming, N.; Pines, O. Class-I and Class-II fumarase enzymes and associated metabolites signal the DNA Damage Response differently during evolution. bioRxiv 2020. [Google Scholar] [CrossRef]
- Singer, E.; Silas, Y.B.; Ben-Yehuda, S.; Pines, O. Bacterial fumarase and L-malic acid are evolutionary ancient components of the DNA damage response. eLife 2017, 6, e30927. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Yu, J.; Wu, J.; Liu, H.; Lin, K.; Zhang, Y. Identification of pathogens causing alfalfa Fusarium root rot in Inner Mongolia, China. Agronomy 2023, 13, 456. [Google Scholar] [CrossRef]
- Gao, Y.; Xiong, X.; Wang, H.; Wang, J.; Bi, Y.; Yan, Y.; Cao, Z.; Li, D.; Song, F. Ero1-Pdi1 module-catalyzed dimerization of a nucleotide sugar transporter, FonNst2, regulates virulence of Fusarium oxysporum on watermelon. Environ. Microbiol. 2022, 3, 1200–1220. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Lou, J.; Wang, J.; Zeng, S.; Xiong, X.; Guo, M.; Li, D.; Song, F. The secreted protein FonCHRD is essential for vegetative growth, asexual reproduction, and pathogenicity in watermelon Fusarium wilt fungus. Crop Health 2024, 2, 16. [Google Scholar] [CrossRef]
- Gao, Y.; Xiong, X.; Wang, H.; Bi, Y.; Wang, J.; Yan, Y.; Li, D.; Song, F. Fusarium oxysporum f. sp. niveum Pumilio 1 regulates virulence on watermelon through Interacting with the ARP2/3 complex and binding to an A-Rich motif in the 3′ UTR of diverse transcripts. mBio 2023, 14, e15723. [Google Scholar] [CrossRef]
- Bi, Y.; Wang, H.; Yuan, X.; Yan, Y.; Li, D.; Song, F. The NAC transcription factor ONAC083 negatively regulates rice immunity against Magnaporthe oryzae by directly activating transcription of the RING-H2 gene OsRFPH2-6. J. Integr. Plant Biol. 2022, 65, 854–875. [Google Scholar] [CrossRef]
- Huang, Z.; Lou, J.; Gao, Y.; Noman, M.; Li, D.; Song, F. FonTup1 functions in growth, conidiogenesis and pathogenicity of Fusarium oxysporum f. sp. niveum through modulating the expression of the tricarboxylic acid cycle genes. Microbiol. Res. 2023, 272, 127389. [Google Scholar] [CrossRef]
- Sun, H.; Cai, S.; Liu, H.; Li, X.; Deng, Y.; Yang, X.; Cao, S.; Li, W.; Chen, H. FgSdhC paralog confers natural resistance toward SDHI fungicides in Fusarium graminearum. J. Agric. Food Chem. 2023, 71, 20643–20653. [Google Scholar] [CrossRef]
- Lushchak, V.I.; Matviishyn, T.M.; Husak, V.V.; Storey, J.M.; Storey, K.B. Pesticide toxicity: A mechanistic approach. EXCLI J. 2018, 17, 1101–1136. [Google Scholar]
- García, P.C.; Rivero, R.M.; Romero, R.L. The role of fungicides in the physiology of higher plants: Implications for defense responses. Bot. Rev. 2003, 69, 162–172. [Google Scholar] [CrossRef]
- Russell, T.M.; Azad, M.G.; Richardson, D.R. The relationship of Glutathione-S-Transferase and multi-drug resistance-related protein 1 in Nitric Oxide (NO) transport and storage. Molecules 2021, 26, 5784. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, M.; Zhou, Z.; Li, J.; Chen, D.; Duan, Y.; Zhou, M. Impact of five succinate dehydrogenase inhibitors on DON biosynthesis of Fusarium asiaticum, Causing Fusarium fead blight in wheat. Toxins 2019, 11, 272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ye, Z.; Yang, S.; Zhang, M.; Xia, Y.; Liu, Z.; Sun, Y.; Li, J.; Li, X.; Zhu, Z.; et al. Succinate dehydrogenase governs the sensitivity to SDHI fungicides and fruit pathogenicity of Colletotrichum gloeosporioides. J. Agric. Food Res. 2025, 19, 101587. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Li, H.; Liu, Y.; Wang, Y.; Xue, J.; Wang, Y.; Wu, Z. FpFumB Is Required for Basic Biological Processes and Virulence in Fusarium proliferatum by Modulating DNA Repair Through Interaction with FpSae2. Microorganisms 2025, 13, 1433. https://doi.org/10.3390/microorganisms13061433
Gao Y, Li H, Liu Y, Wang Y, Xue J, Wang Y, Wu Z. FpFumB Is Required for Basic Biological Processes and Virulence in Fusarium proliferatum by Modulating DNA Repair Through Interaction with FpSae2. Microorganisms. 2025; 13(6):1433. https://doi.org/10.3390/microorganisms13061433
Chicago/Turabian StyleGao, Yizhou, Haibo Li, Yong Liu, Yuqing Wang, Jingwen Xue, Yitong Wang, and Zhihong Wu. 2025. "FpFumB Is Required for Basic Biological Processes and Virulence in Fusarium proliferatum by Modulating DNA Repair Through Interaction with FpSae2" Microorganisms 13, no. 6: 1433. https://doi.org/10.3390/microorganisms13061433
APA StyleGao, Y., Li, H., Liu, Y., Wang, Y., Xue, J., Wang, Y., & Wu, Z. (2025). FpFumB Is Required for Basic Biological Processes and Virulence in Fusarium proliferatum by Modulating DNA Repair Through Interaction with FpSae2. Microorganisms, 13(6), 1433. https://doi.org/10.3390/microorganisms13061433