
Host-Determined Diversity and Environment-Shaped Community Assembly of Phyllosphere Microbiomes in Alpine Steppes Ecosystems

Abstract
1. Introduction
2. Materials and Methods
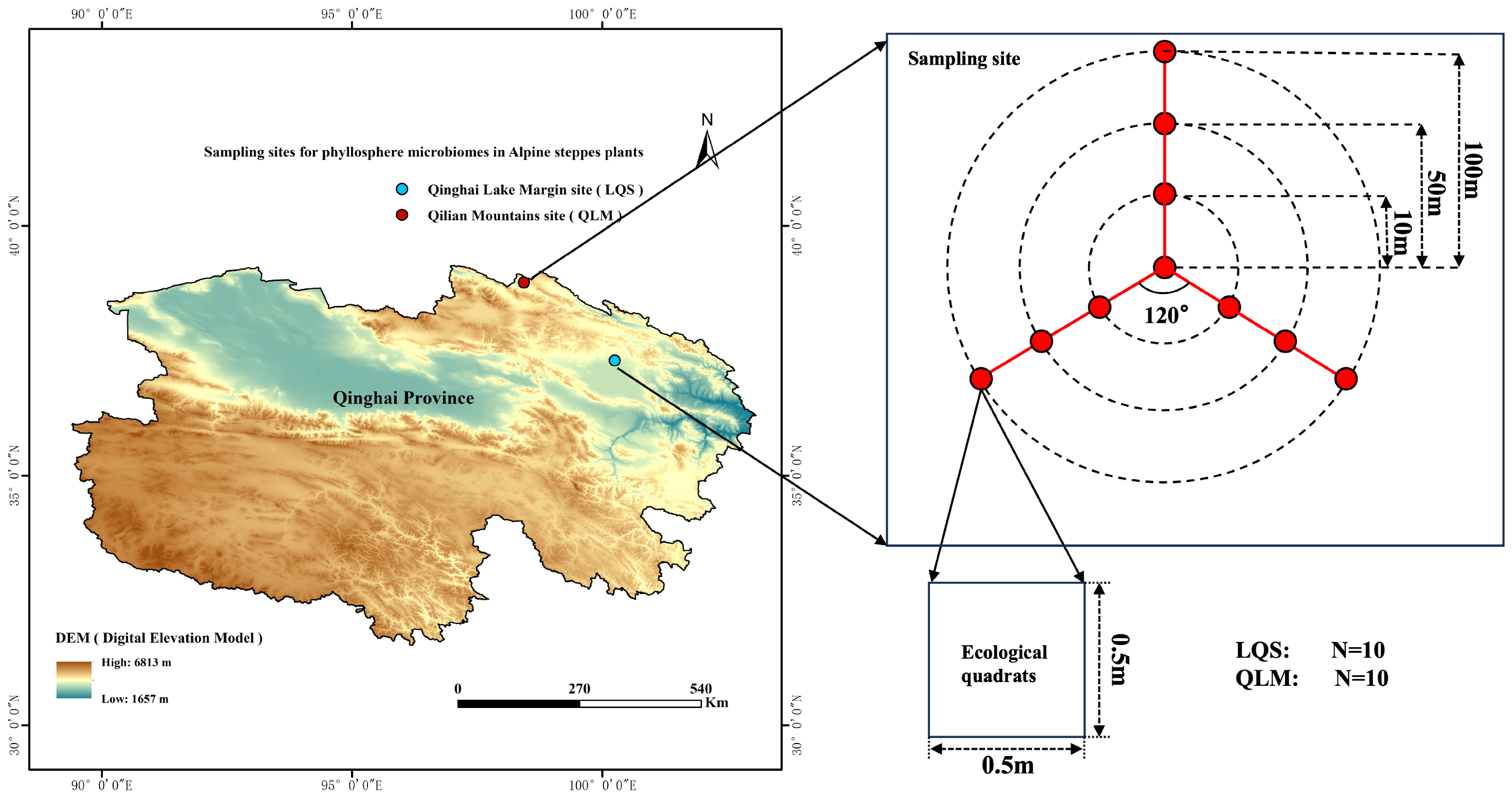
2.1. Study Area Characteristics
2.2. Site Layout, Vegetation Survey, Phyllosphere Sampling and Preservation, and Soil Sampling
2.3. Phyllosphere Microbial Sample Processing, Sequencing, and Bioinformatic Analysis
2.3.1. Microbial Elution from Phyllosphere Samples
2.3.2. DNA Extraction and Quality Assessment
2.3.3. PCR Amplification of 16S rRNA and ITS Regions
2.3.4. PCR Product Verification, Purification, and Sequencing
2.3.5. Sequence Quality Control and OTU Clustering
2.3.6. OTU Assignment and Taxonomic Annotation
2.4. Statistical Analysis
3. Results
3.1. Taxonomic Composition of Phyllosphere Microbial Communities
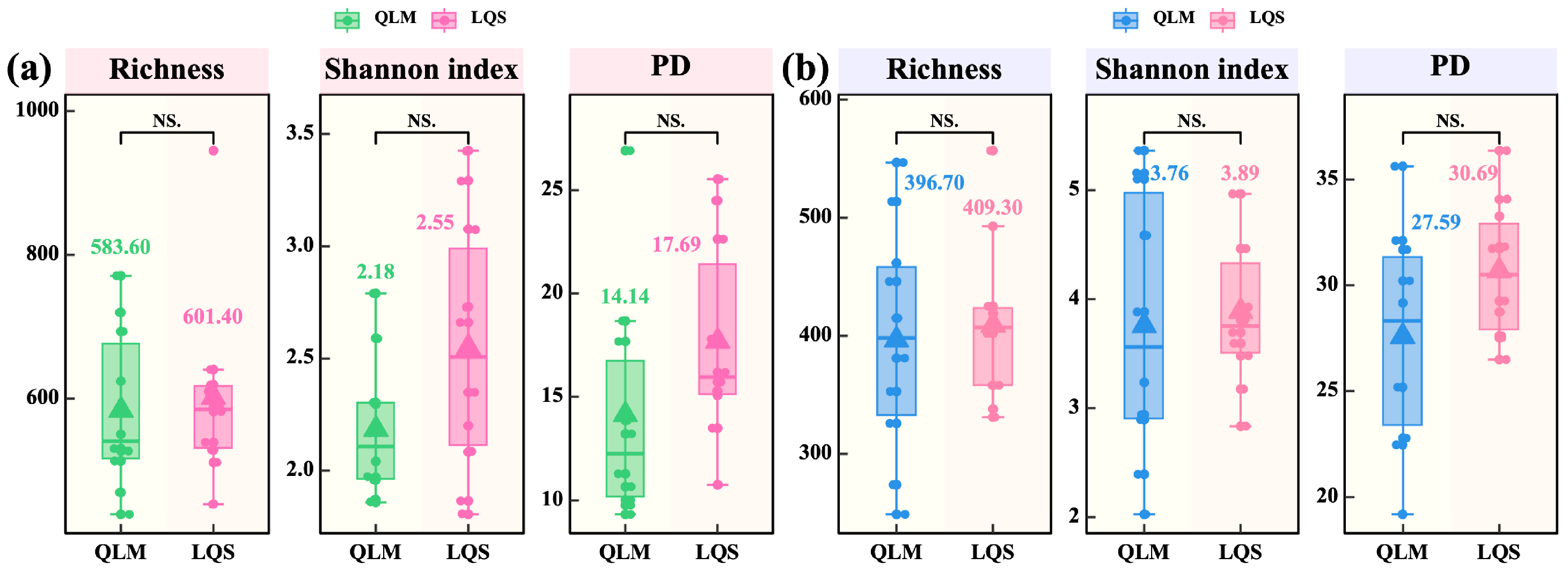
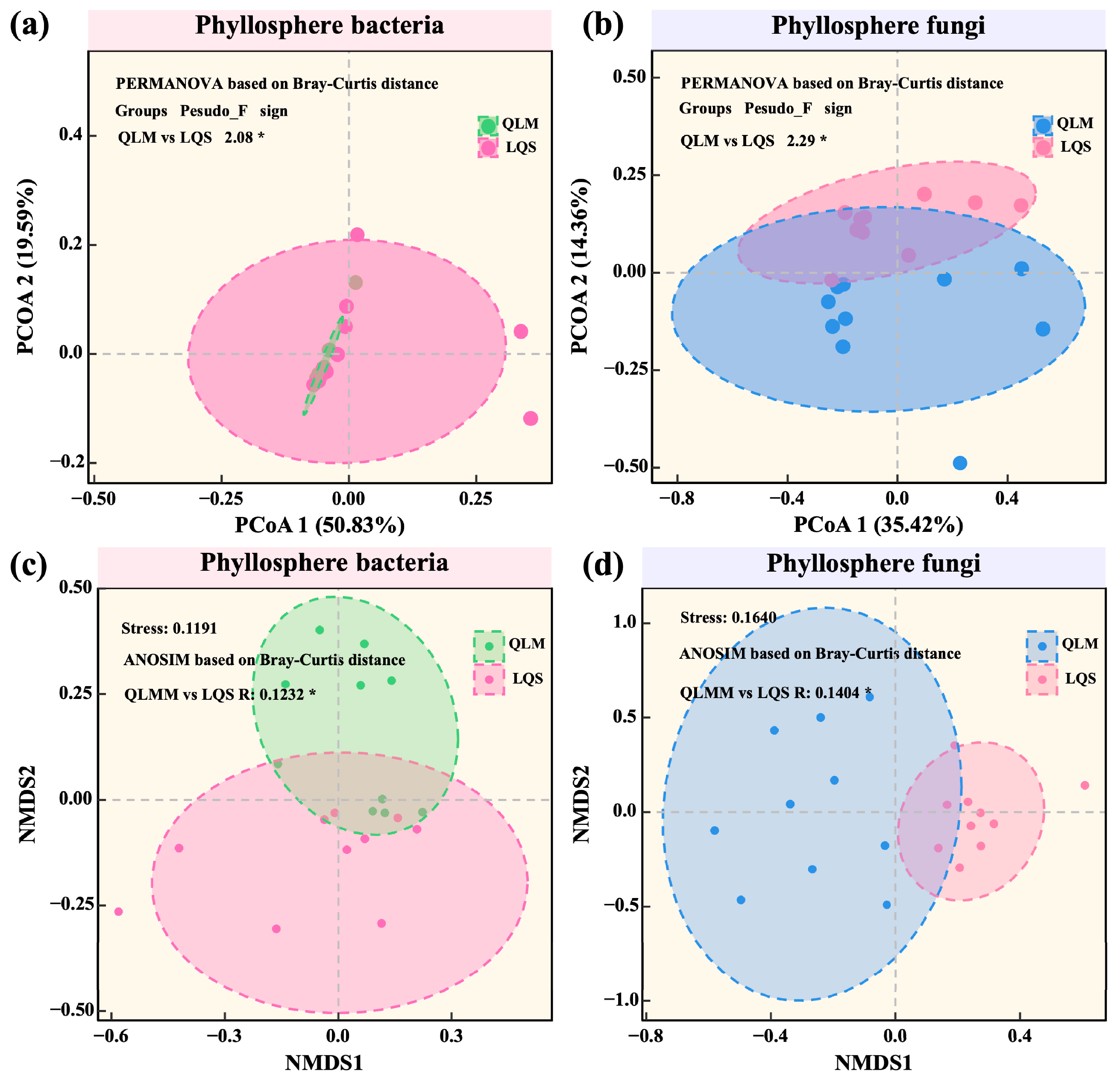
3.2. Diversity, Community Structure, and Environmental Drivers of Phyllosphere Microbiota
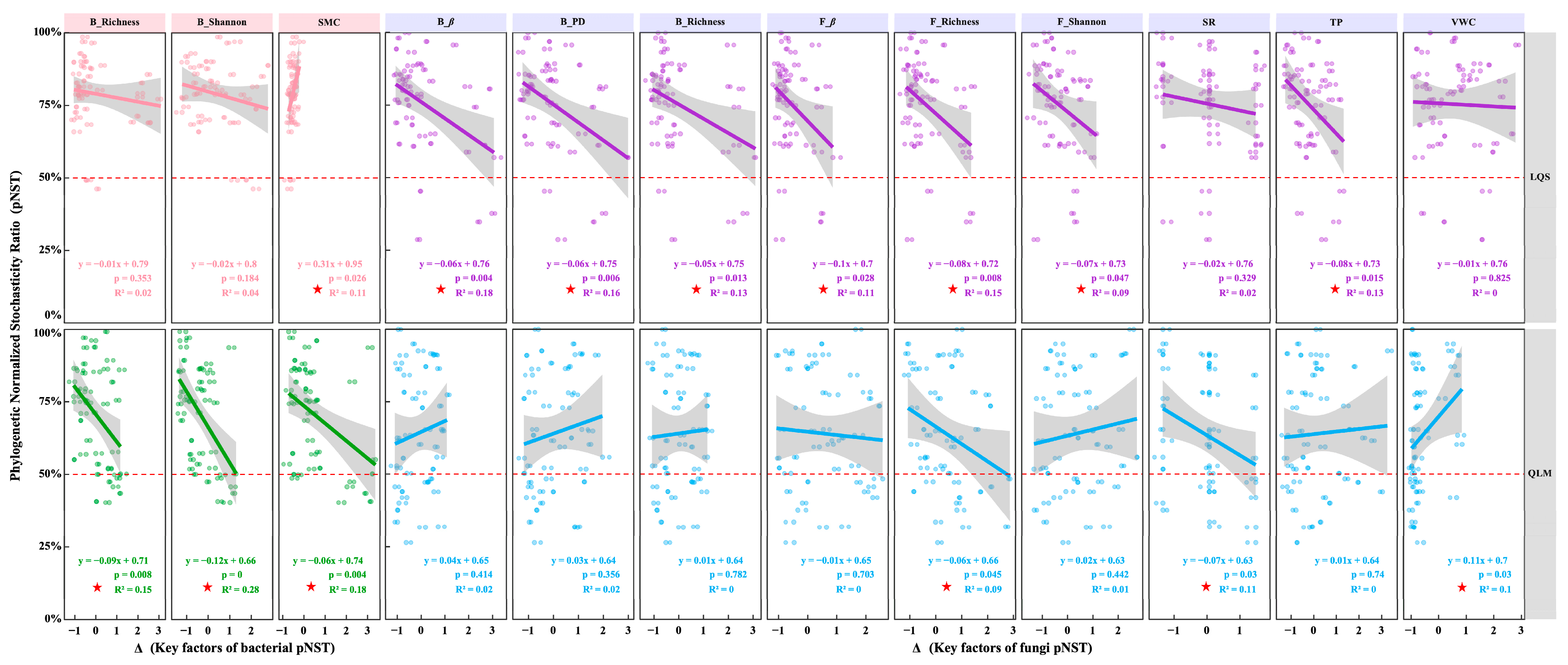
3.3. Host and Environmental Factors Shape Assembly of Phyllosphere Microbial Communities
4. Discussion
4.1. Regional Divergence of Alpine Phyllosphere Microbiota
4.2. Host–Environment Interplay Shapes Phyllosphere Diversity Patterns in Alpine Steppes
4.3. Host and Environmental Drivers of Phyllosphere Community Assembly
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.B.; Kent, J. Biodiversity Hotspots for Conservation Priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhu, Q.; Peng, C.; Wu, N.; Wang, Y.; Fang, X.; Gao, Y.; Zhu, D.; Yang, G.; Tian, J.; et al. The Impacts of Climate Change and Human Activities on Biogeochemical Cycles on the Qinghai-Tibetan Plateau. Glob. Change Biol. 2013, 19, 2940–2955. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.; Dong, J.; Xiao, X. Green-up Dates in the Tibetan Plateau Have Continuously Advanced from 1982 to 2011. Proc. Natl. Acad. Sci. USA 2013, 110, 4309–4314. [Google Scholar] [CrossRef]
- Li, L.; Chen, J.; Han, X.; Zhang, W.; Shao, C. Alpine Steppe Ecosystem. In Grassland Ecosystems of China: A Synthesis and Resume; Li, L., Chen, J., Han, X., Zhang, W., Shao, C., Eds.; Springer: Singapore, 2020; pp. 285–306. ISBN 978-981-15-3421-8. [Google Scholar]
- Hao, A.; Duan, H.; Wang, X.; Zhao, G.; You, Q.; Peng, F.; Du, H.; Liu, F.; Li, C.; Lai, C.; et al. Different Response of Alpine Meadow and Alpine Steppe to Climatic and Anthropogenic Disturbance on the Qinghai-Tibetan Plateau. Glob. Ecol. Conserv. 2021, 27, e01512. [Google Scholar] [CrossRef]
- Jin, X.; Deng, A.; Fan, Y.; Ma, K.; Zhao, Y.; Wang, Y.; Zheng, K.; Zhou, X.; Lu, G. Diversity, Functionality, and Stability: Shaping Ecosystem Multifunctionality in the Successional Sequences of Alpine Meadows and Alpine Steppes on the Qinghai-Tibet Plateau. Front. Plant Sci. 2025, 16, 1436439. [Google Scholar] [CrossRef]
- Wang, W.; Li, S.; Degen, A.A.; Gauchan, D.; Shang, Z. Chapter 4—Alpine Steppe Degradation: Status, Causes, and Trends. In Grassland Degradation, Restoration and Sustainable Management of Global Alpine Area; Shang, Z., Degen, A.A., Dong, S., Zhou, H., Ma, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2025; pp. 87–102. ISBN 978-0-443-21882-8. [Google Scholar]
- Zhou, H.; Zhang, D.; Jiang, Z.; Sun, P.; Xiao, H.; Yuxin, W.; Chen, J. Changes in the Soil Microbial Communities of Alpine Steppe at Qinghai-Tibetan Plateau under Different Degradation Levels. Sci. Total Environ. 2019, 651, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Xu-Ri; Liu, Y.; Wang, Y.; Wang, Y. Three-Year Study of CO2 Efflux and CH4/N2O Fluxes at an Alpine Steppe Site on the Central Tibetan Plateau and Their Responses to Simulated N Deposition. Geoderma 2014, 232–234, 88–96. [Google Scholar] [CrossRef]
- Zhu, Y.-G.; Xiong, C.; Wei, Z.; Chen, Q.-L.; Ma, B.; Zhou, S.-Y.-D.; Tan, J.; Zhang, L.-M.; Cui, H.-L.; Duan, G.-L. Impacts of Global Change on the Phyllosphere Microbiome. New Phytol. 2022, 234, 1977–1986. [Google Scholar] [CrossRef]
- Remus-Emsermann, M.N.P.; Lücker, S.; Müller, D.B.; Potthoff, E.; Daims, H.; Vorholt, J.A. Spatial Distribution Analyses of Natural Phyllosphere-Colonizing Bacteria on Rabidopsis Thaliana Revealed by Fluorescence in Situ Hybridization. Environ. Microbiol. 2014, 16, 2329–2340. [Google Scholar] [CrossRef]
- Vorholt, J.A. Microbial Life in the Phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef]
- Remus-Emsermann, M.N.P.; Schlechter, R.O. Phyllosphere Microbiology: At the Interface between Microbial Individuals and the Plant Host. New Phytol. 2018, 218, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Sahu, K.P.; Patel, A.; Kumar, M.; Sheoran, N.; Mehta, S.; Reddy, B.; Eke, P.; Prabhakaran, N.; Kumar, A. Integrated Metabarcoding and Culturomic-Based Microbiome Profiling of Rice Phyllosphere Reveal Diverse and Functional Bacterial Communities for Blast Disease Suppression. Front. Microbiol. 2021, 12, 780458. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jin, M.-K.; Neilson, R.; Hu, S.-L.; Tang, Y.-J.; Zhang, Z.; Huang, F.-Y.; Zhang, J.; Yang, X.-R. Plant Identity Shapes Phyllosphere Microbiome Structure and Abundance of Genes Involved in Nutrient Cycling. Sci. Total Environ. 2023, 865, 161245. [Google Scholar] [CrossRef]
- Yang, X.; Dai, Z.; Yuan, R.; Guo, Z.; Xi, H.; He, Z.; Wei, M. Effects of Salinity on Assembly Characteristics and Function of Microbial Communities in the Phyllosphere and Rhizosphere of Salt-Tolerant Avicennia Marina Mangrove Species. Microbiol. Spectrum 2023, 11, e0300022. [Google Scholar] [CrossRef]
- Kong, X.; Han, Z.; Tai, X.; Jin, D.; Ai, S.; Zheng, X.; Bai, Z. Maize (Zea mays L. sp.) Varieties Significantly Influence Bacterial and Fungal Community in Bulk Soil, Rhizosphere Soil and Phyllosphere. FEMS Microbiol. Ecol. 2020, 96, fiaa020. [Google Scholar] [CrossRef]
- Huang, S.; Zha, X.; Fu, G. Affecting Factors of Plant Phyllosphere Microbial Community and Their Responses to Climatic Warming—A Review. Plants 2023, 12, 2891. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Zhang, H.; Liu, Y.; Chen, J.; Li, J. Drought-Resistant Trait of Different Crop Genotypes Determines Assembly Patterns of Soil and Phyllosphere Microbial Communities. Microbiol. Spectrum 2023, 11, e00068-23. [Google Scholar] [CrossRef]
- Finkel, O.M.; Burch, A.Y.; Lindow, S.E.; Post, A.F.; Belkin, S. Geographical Location Determines the Population Structure in Phyllosphere Microbial Communities of a Salt-Excreting Desert Tree. Appl. Environ. Microbiol. 2011, 77, 7647–7655. [Google Scholar] [CrossRef]
- Cui, X.; Tang, Y.; Gu, S.; Nishimura, S.; Shi, S.; Zhao, X. Photosynthetic Depression in Relation to Plant Architecture in Two Alpine Herbaceous Species. Environ. Exp. Bot. 2003, 50, 125–135. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Guo, Q.; Yao, W. Molecular Characterisation of a DREB Gene from Sophora moorcroftiana, an Endemic Species of Plateau. Protoplasma 2017, 254, 1735–1741. [Google Scholar] [CrossRef]
- Fan, Z.; Bai, X. Scenarios of Potential Vegetation Distribution in the Different Gradient Zones of Qinghai-Tibet Plateau under Future Climate Change. Sci. Total Environ. 2021, 796, 148918. [Google Scholar] [CrossRef]
- Wang, Y.; Lü, Y.; Lü, D.; Yin, L.; Wang, X. Climate Change and Its Ecological Risks Are Spatially Heterogeneous in High-Altitude Region: The Case of Qinghai-Tibet Plateau. Catena 2024, 243, 108140. [Google Scholar] [CrossRef]
- Wu, M.-H.; Xue, K.; Wei, P.-J.; Jia, Y.-L.; Zhang, Y.; Chen, S.-Y. Soil Microbial Distribution and Assembly Are Related to Vegetation Biomass in the Alpine Permafrost Regions of the Qinghai-Tibet Plateau. Sci. Total Environ. 2022, 834, 155259. [Google Scholar] [CrossRef]
- Wang, S.; Jiao, C.; Zhao, D.; Zeng, J.; Xing, P.; Liu, Y.; Wu, Q.L. Disentangling the Assembly Mechanisms of Bacterial Communities in a Transition Zone between the Alpine Steppe and Alpine Meadow Ecosystems on the Tibetan Plateau. Sci. Total Environ. 2022, 847, 157446. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, K.; Tao, K.; Hou, T. Biogeographical Patterns and Drivers of Bacterial Community in the Qinghai-Tibetan Plateau. Appl. Soil Ecol. 2023, 183, 104757. [Google Scholar] [CrossRef]
- Duan, H.; Xue, X.; Wang, T.; Kang, W.; Liao, J.; Liu, S. Spatial and Temporal Differences in Alpine Meadow, Alpine Steppe and All Vegetation of the Qinghai-Tibetan Plateau and Their Responses to Climate Change. Remote Sens. 2021, 13, 669. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, S.; Liu, C.; Sun, J.; Liu, Y.; Wang, F. Interactive Response and Spatial Heterogeneity of Alpine Grassland Biodiversity to Soil, Topography, Climate, and Human Factors on the Qinghai-Tibet Plateau. Land Degrad. Dev. 2023, 34, 4488–4502. [Google Scholar] [CrossRef]
- Smets, W.; Spada, L.M.; Gandolfi, I.; Wuyts, K.; Legein, M.; Muyshondt, B.; Samson, R.; Franzetti, A.; Lebeer, S. Bacterial Succession and Community Dynamics of the Emerging Leaf Phyllosphere in Spring. Microbiol. Spectrum 2022, 10, e02420-21. [Google Scholar] [CrossRef]
- Liu, S.; Chou, M.-Y.; Benucci, G.M.N.; Eudes, A.; Bonito, G. Genetic Modification of the Shikimate Pathway to Reduce Lignin Content in Switchgrass (Panicum virgatum L.) Significantly Impacts Plant Microbiomes. Microbiol. Spectrum 2024, 13, e01546-24. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E.; Goebel, B.M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S rRNA Sequence Analysis in the Present Species Definition in Bacteriology. Int. J. Syst. Evol. Microbiol. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Faith, D.P. Conservation Evaluation and Phylogenetic Diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a Package of R Functions for Community Ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Anderson, M.J. A New Method for Non-Parametric Multivariate Analysis of Variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Liu, Y.-X.; Chen, L.; Ma, T.; Li, X.; Zheng, M.; Zhou, X.; Chen, L.; Qian, X.; Xi, J.; Lu, H.; et al. EasyAmplicon: An Easy-to-Use, Open-Source, Reproducible, and Community-Based Pipeline for Amplicon Data Analysis in Microbiome Research. Imeta 2023, 2, e83. [Google Scholar] [CrossRef] [PubMed]
- Ning, D.; Deng, Y.; Tiedje, J.M.; Zhou, J. A General Framework for Quantitatively Assessing Ecological Stochasticity. Proc. Natl. Acad. Sci. USA 2019, 116, 16892–16898. [Google Scholar] [CrossRef]
- Stone, B.W.G.; Jackson, C.R. Seasonal Patterns Contribute More towards Phyllosphere Bacterial Community Structure than Short-Term Perturbations. Microb. Ecol. 2021, 81, 146–156. [Google Scholar] [CrossRef]
- Rastogi, G.; Coaker, G.L.; Leveau, J.H.J. New Insights into the Structure and Function of Phyllosphere Microbiota through High-Throughput Molecular Approaches. FEMS Microbiol. Lett. 2013, 348, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-P.; Zhang, M.-X.; Lü, Z.-L.; He, Y.-Y.; Yang, X.-X.; Khan, A.; Xiong, Y.-C.; Fang, X.-L.; Dong, Q.-M.; Zhang, J.-L. Grazing Practices Affect Phyllosphere and Rhizosphere Bacterial Communities of Kobresia humilis by Altering Their Network Stability. Sci. Total Environ. 2023, 900, 165814. [Google Scholar] [CrossRef] [PubMed]
- Bataeva, Y.V.; Grigoryan, L.N. Ecological Features and Adaptive Capabilities of Cyanobacteria in Desert Ecosystems: A Review. Eurasian Soil Sci. 2024, 57, 430–445. [Google Scholar] [CrossRef]
- Thapa, S.; Prasanna, R.; Ranjan, K.; Velmourougane, K.; Ramakrishnan, B. Nutrients and Host Attributes Modulate the Abundance and Functional Traits of Phyllosphere Microbiome in Rice. Microbiol. Res. 2017, 204, 55–64. [Google Scholar] [CrossRef]
- Wang, X.; Yuan, Z.; Ali, A.; Yang, T.; Lin, F.; Mao, Z.; Ye, J.; Fang, S.; Hao, Z.; Wang, X.; et al. Leaf Traits and Temperature Shape the Elevational Patterns of Phyllosphere Microbiome. J. Biogeogr. 2023, 50, 2135–2147. [Google Scholar] [CrossRef]
- Laforest-Lapointe, I.; Messier, C.; Kembel, S.W. Host Species Identity, Site and Time Drive Temperate Tree Phyllosphere Bacterial Community Structure. Microbiome 2016, 4, 27. [Google Scholar] [CrossRef]
- Nekola, J.C.; White, P.S. Special Paper: The Distance Decay of Similarity in Biogeography and Ecology. J. Biogeogr. 1999, 26, 867–878. [Google Scholar] [CrossRef]
- Li, M.; Hong, L.; Ye, W.; Wang, Z.; Shen, H. Phyllosphere Bacterial and Fungal Communities Vary with Host Species Identity, Plant Traits and Seasonality in a Subtropical Forest. Environ. Microbiome 2022, 17, 29. [Google Scholar] [CrossRef]
- Qu, X.; Pan, Y.; Wang, P.; Ran, L.; Qin, G.; Li, Q.; Kang, P. Response of Phyllosphere and Rhizosphere Microbial Communities to Salt Stress of Tamarix Chinensis. Plants 2024, 13, 1091. [Google Scholar] [CrossRef]
- Zhou, J.; Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002-17. [Google Scholar] [CrossRef]
- Qu, S.; Shen, C.; Zhang, L.; Wang, J.; Zhang, L.-M.; Chen, B.; Sun, G.-X.; Ge, Y. Dispersal Limitation and Host Selection Drive Geo-Specific and Plant-Specific Differentiation of Soil Bacterial Communities in the Tibetan Alpine Ecosystem. Sci. Total Environ. 2023, 863, 160944. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Caruso, T.; Archer, S.D.J.; Gillman, L.N.; Lau, M.C.Y.; Cary, S.C.; Lee, C.K.; Pointing, S.B. Stochastic and Deterministic Effects of a Moisture Gradient on Soil Microbial Communities in the McMurdo Dry Valleys of Antarctica. Front. Microbiol. 2018, 9, 2619. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, G.; Zhang, Z.; Xia, S.; Yang, X. Soil Total Phosphorus Mediate the Assembly Processes of Rhizosphere Microbial Communities of Ficus Species in a Tropical Rainforest. Rhizosphere 2024, 32, 100986. [Google Scholar] [CrossRef]
- Daniel, J.; Gleason, J.E.; Cottenie, K.; Rooney, R.C. Stochastic and Deterministic Processes Drive Wetland Community Assembly across a Gradient of Environmental Filtering. Oikos 2019, 128, 1158–1169. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environmental and Plant | Factors | Qinghai Lake Margin Site (LQS) | Qilian Mountains Site (QLM) |
|---|---|---|---|
| Climatic Factors | MAT (mm) | 0.61 ± 0.03 a | −3.91 ± 0.06 b |
| MAP (°C) | 457.28 ± 0.26 a | 330.92 ± 1.53 b | |
| Plant Traits | Biomass (g/m2) | 61.44 ± 5.40 a | 52.41 ± 2.37 a |
| Species richness | 5.11 ± 0.88 a | 4.20 ± 0.79 b | |
| Height (cm) | 6.42 ± 1.52 b | 8.24 ± 1.73 a | |
| Cover (%) | 86.41 ± 5.04 a | 80.7 ± 6.62 a | |
| VWC (g/m2) | 9.44 + 2.35 a | 2.82 ± 1.95 b | |
| TN (mg/kg) | 2175.97 ± 412.97 a | 2097.32 ± 258.84 a | |
| TP (mg/kg) | 295.27 ± 40.11 a | 335.46 ± 59.17 a | |
| OM (%) | 5.01 ± 0.67 a | 4.16 ± 0.63 b | |
| Soil | NH4+-N (mg/kg) | 5.46 ± 1.96 b | 8.18 ± 2.25 a |
| Physicochemical | NO3−-N (mg/kg) | 22.39 ± 2.23 a | 31.07 ± 4.87 a |
| Properties | AP (mg/kg) | 6.71 ± 2.35 a | 6.11 ± 2.14 a |
| AK (mg/kg) | 202.78 ± 84.88 a | 197.73 ± 26.82 a | |
| SMC (%) | 36.15 ± 3.75 a | 33.38 ± 7.48 a | |
| EC (us/cm) | 0.38 ± 0.11 a | 0.99 ± 0.24 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, K.; Jin, X.; Li, J.; Lu, G. Host-Determined Diversity and Environment-Shaped Community Assembly of Phyllosphere Microbiomes in Alpine Steppes Ecosystems. Microorganisms 2025, 13, 1432. https://doi.org/10.3390/microorganisms13061432
Zheng K, Jin X, Li J, Lu G. Host-Determined Diversity and Environment-Shaped Community Assembly of Phyllosphere Microbiomes in Alpine Steppes Ecosystems. Microorganisms. 2025; 13(6):1432. https://doi.org/10.3390/microorganisms13061432
Chicago/Turabian StyleZheng, Kaifu, Xin Jin, Jingjing Li, and Guangxin Lu. 2025. "Host-Determined Diversity and Environment-Shaped Community Assembly of Phyllosphere Microbiomes in Alpine Steppes Ecosystems" Microorganisms 13, no. 6: 1432. https://doi.org/10.3390/microorganisms13061432
APA StyleZheng, K., Jin, X., Li, J., & Lu, G. (2025). Host-Determined Diversity and Environment-Shaped Community Assembly of Phyllosphere Microbiomes in Alpine Steppes Ecosystems. Microorganisms, 13(6), 1432. https://doi.org/10.3390/microorganisms13061432