
Heterologous Expression of the Nitrogen-Fixing Gene Cluster from Paenibacillus polymyxa in Bacillus subtilis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Growth Conditions
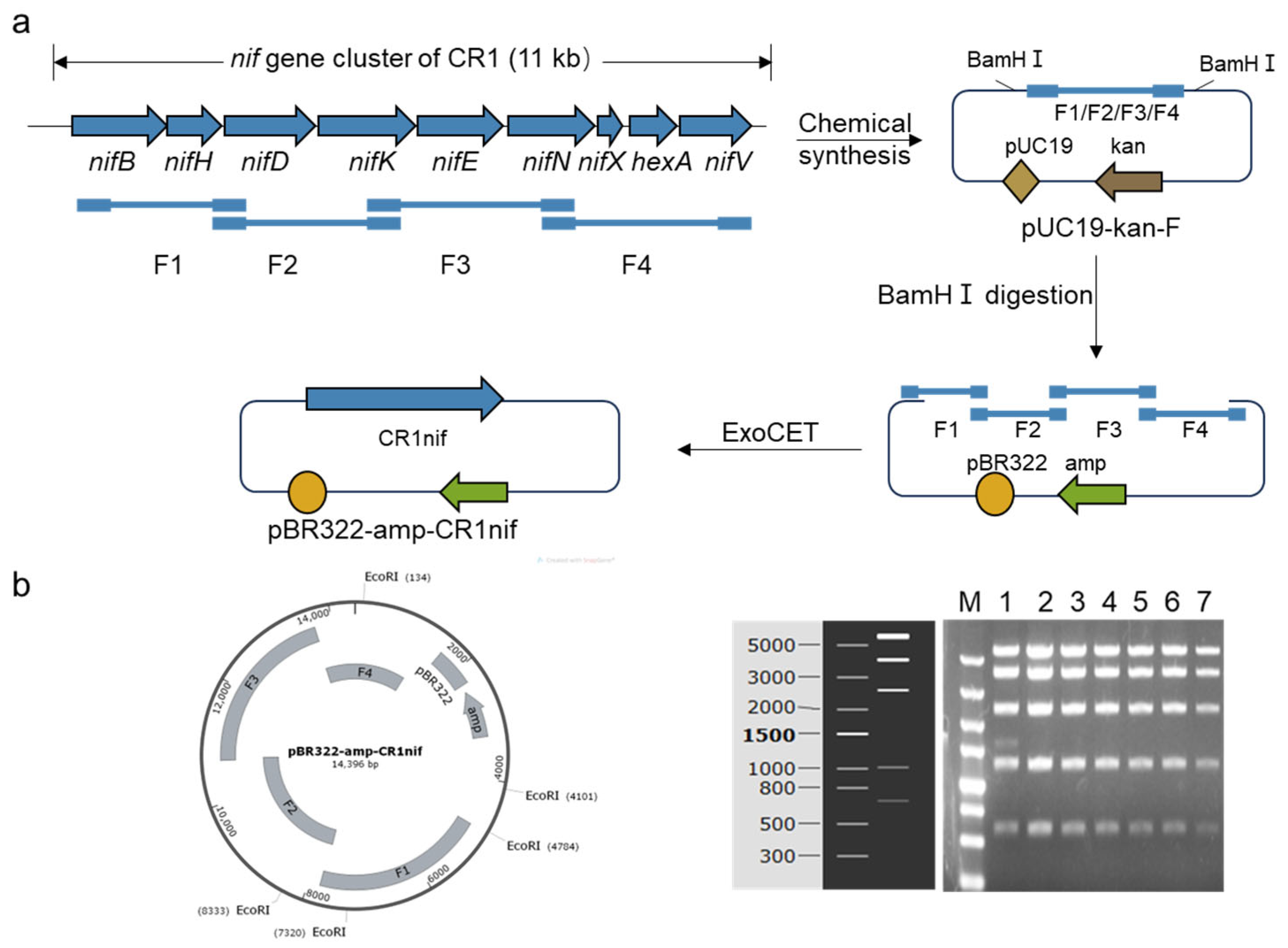
2.2. Assembly of the nif Gene Cluster from P. polymyxa CR1
2.3. Integration of the nif Gene Cluster into the Genome of B. subtilis 168
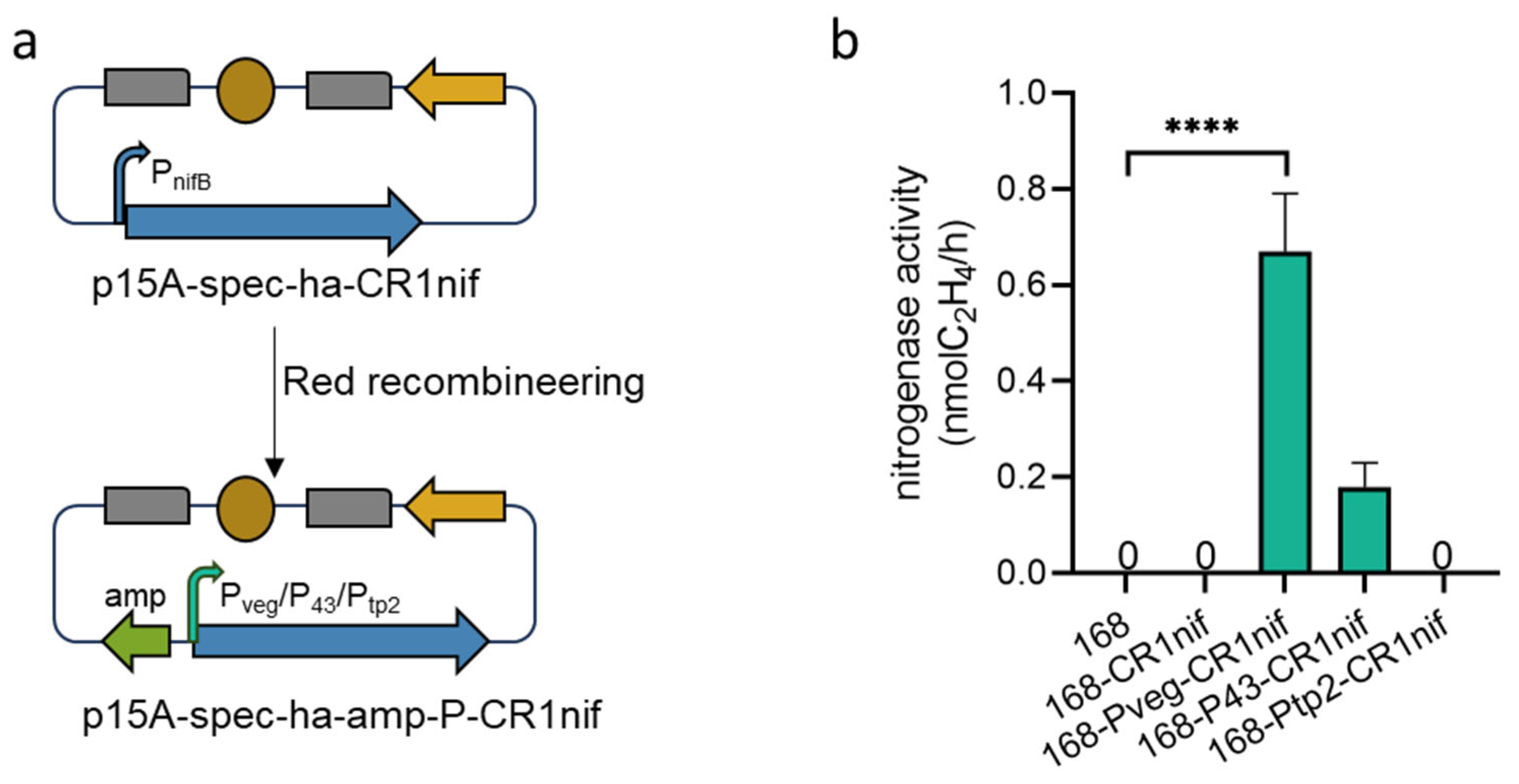
2.4. Substitution of the nifB Promoter
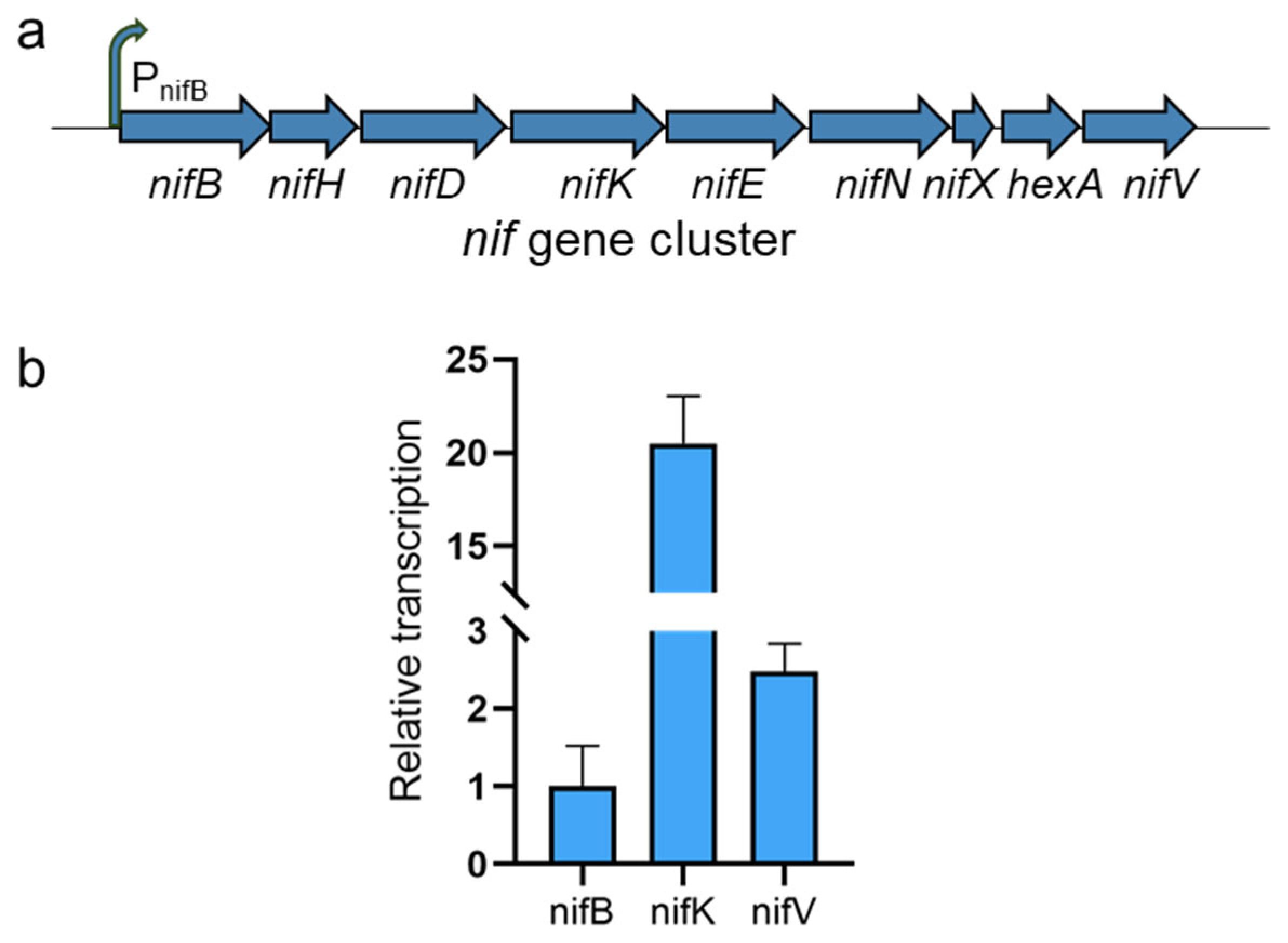
2.5. Reverse Transcription Quantitative PCR (RT-qPCR) Analysis
2.6. Acetylene Reduction Assays
3. Results and Discussion
3.1. The Assembly of the Nitrogen-Fixing Gene Cluster from P. polymyxa CR1
3.2. Integration of the Nitrogen-Fixing Gene Cluster from P. polymyxa CR1 into B. subtilis 168
3.3. Transcription of the nif Gene Cluster in B. subtilis
3.4. Heterologous Expression of the nif Gene Cluster in B. subtilis 168
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ExoCET | Exonuclease combined with RecET recombination |
| ARA | Acetylene reduction assay |
| PGPR | Plant-growth-promoting rhizobacterium |
| RT-qPCR | Real-time quantitative reverse transcription |
References
- Rucker, H.R.; Kaçar, B. Enigmatic evolution of microbial nitrogen fixation: Insights from Earth’s past. Trends Microbiol. 2024, 32, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sindhu, S.S.; Kumar, R. Biofertilizers: An ecofriendly technology for nutrient recycling and environmental sustainability. Curr. Res. Microb. Sci. 2022, 3, 100094. [Google Scholar] [CrossRef] [PubMed]
- Ladha, J.K.; Peoples, M.B.; Reddy, P.M.; Biswas, J.C.; Bennett, A.; Jat, M.L.; Krupnik, T.J. Biological nitrogen fixation and prospects for ecological intensification in cereal-based cropping systems. Field Crops Res. 2022, 283, 108541. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sangwan, S.; Kaur, H.; Patra, A.; Anamika; Mehta, S. Diversity and evolution of nitrogen fixing bacteria. In Sustainable Agriculture Reviews 60: Microbial Processes in Agriculture; Singh, N.K., Chattopadhyay, A., Lichtfouse, E., Eds.; Springer Nature: Cham, Switzerland, 2023; pp. 95–120. [Google Scholar]
- Goyal, R.K.; Mattoo, A.K.; Schmidt, M.A. Rhizobial-host interactions and symbiotic nitrogen fixation in legume crops toward agriculture sustainability. Front. Microbiol. 2021, 12, 669404. [Google Scholar] [CrossRef]
- Quechol, R.; Solomon, J.B.; Liu, Y.A.; Lee, C.C.; Jasniewski, A.J.; Górecki, K.; Oyala, P.; Hedman, B.; Hodgson, K.O.; Ribbe, M.W.; et al. Heterologous synthesis of the complex homometallic cores of nitrogenase P- and M-clusters in Escherichia coli. Proc. Natl. Acad. Sci. USA 2023, 120, e2314788120. [Google Scholar] [CrossRef]
- Bennett, E.M.; Murray, J.W.; Isalan, M. Engineering nitrogenases for synthetic nitrogen fixation: From pathway engineering to directed evolution. BioDes. Res. 2023, 5, 0005. [Google Scholar] [CrossRef]
- Solomon, J.B.; Lee, C.C.; Liu, Y.A.; Duffin, C.; Ribbe, M.W.; Hu, Y. Ammonia synthesis via an engineered nitrogenase assembly pathway in Escherichia coli. Nat. Catal. 2024, 7, 1130–1141. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; Liu, Z.; Zhao, D.; Liu, X.; Zhang, B.; Xie, J.; Hong, Y.; Li, P.; Chen, S.; et al. A minimal nitrogen fixation gene cluster from Paenibacillus sp. WLY78 enables expression of active nitrogenase in Escherichia coli. PLoS Genet. 2013, 9, e1003865. [Google Scholar] [CrossRef]
- Yang, J.; Xie, X.; Xiang, N.; Tian, Z.-X.; Dixon, R.; Wang, Y.-P. Polyprotein strategy for stoichiometric assembly of nitrogen fixation components for synthetic biology. Proc. Natl. Acad. Sci. USA 2018, 115, E8509–E8517. [Google Scholar] [CrossRef]
- Wahab, A.; Batool, F.; Abdi, G.; Muhammad, M.; Ullah, S.; Zaman, W. Role of plant growth-promoting rhizobacteria in sustainable agriculture: Addressing environmental and biological challenges. J. Plant Physiol. 2025, 307, 154455. [Google Scholar] [CrossRef]
- Hashem, A.; Tabassum, B.; Fathi Abd Allah, E. Bacillus subtilis: A plant-growth promoting rhizobacterium that also impacts biotic stress. Saudi J. Biol. Sci. 2019, 26, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pei, Y.; Wang, X.; Dai, X.; Zhu, M. Antimicrobial metabolites produced by the plant growth-promoting rhizobacteria (PGPR): Bacillus and Pseudomonas. Adv. Agrochem. 2024, 3, 206–221. [Google Scholar] [CrossRef]
- Khan, A.R.; Mustafa, A.; Hyder, S.; Valipour, M.; Rizvi, Z.F.; Gondal, A.S.; Yousuf, Z.; Iqbal, R.; Daraz, U. Bacillus spp. as bioagents: Uses and application for sustainable agriculture. Biology 2022, 11, 1763. [Google Scholar] [CrossRef] [PubMed]
- Put, H.; Gerstmans, H.; Vande Capelle, H.; Fauvart, M.; Michiels, J.; Masschelein, J. Bacillus subtilis as a host for natural product discovery and engineering of biosynthetic gene clusters. Nat. Prod. Rep. 2024, 41, 1113–1151. [Google Scholar] [CrossRef]
- Ortiz, A.; Sansinenea, E.; Keswani, C.; Minkina, T.; Singh, S.P.; Rekadwad, B.; Borriss, R.; Hefferon, K.; Hoat, T.X.; Mitra, D.; et al. Bioengineering Bacillus spp. for sustainable crop production: Recent advances and resources for biotechnological applications. J. Plant Growth Regul. 2024, 44, 1868–1885. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.; Jia, R.; Yin, J.; Li, A.; Xia, L.; Yin, Y.; Müller, R.; Fu, J.; Stewart, A.F.; et al. ExoCET: Exonuclease in vitro assembly combined with RecET recombination for highly efficient direct DNA cloning from complex genomes. Nucleic Acids Res. 2018, 46, e28. [Google Scholar] [CrossRef]
- Liu, Q.; Li, R.; Shi, H.; Yang, R.; Shen, Q.; Cui, Q.; Wang, X.; Li, A.; Zhang, Y.; Fu, J. A recombineering system for Bacillus subtilis based on the native phage recombinase pair YqaJ/YqaK. Eng. Microbiol. 2023, 3, 100099. [Google Scholar] [CrossRef]
- Eastman, A.W.; Heinrichs, D.E.; Yuan, Z.-C. Comparative and genetic analysis of the four sequenced Paenibacillus polymyxa genomes reveals a diverse metabolism and conservation of genes relevant to plant-growth promotion and competitiveness. BMC Genom. 2014, 15, 851. [Google Scholar] [CrossRef]
- Zocca, V.F.B.; Corrêa, G.G.; Lins, M.; de Jesus, V.N.; Tavares, L.F.; Amorim, L.; Kundlatsch, G.E.; Pedrolli, D.B. The CRISPR toolbox for the gram-positive model bacterium Bacillus subtilis. Crit. Rev. Biotechnol. 2022, 42, 813–826. [Google Scholar] [CrossRef]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef]
- Li, Q.; Li, Y.; Li, X.; Chen, S. Identification of genes Involved in Fe–S cluster biosynthesis of nitrogenase in Paenibacillus polymyxa WLY78. Int. J. Mol. Sci. 2021, 22, 3771. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Mao, Z.; Guo, J.; Wei, L.; Ma, H.; Tang, Y.; Chen, T.; Wang, Z.; Zhao, X. Construction, model-based analysis, and characterization of a promoter library for fine-tuned gene expression in Bacillus subtilis. ACS Synth Biol. 2018, 7, 1785–1797. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Li, L.; Zhou, L.; Zhang, R.; Xu, Y.; Li, J. Improving expression of bovine lactoferrin N-lobe by promoter optimization and codon engineering in Bacillus subtilis and Its antibacterial activity. J. Agric. Food Chem. 2019, 67, 9749–9756. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Zheng, W.; Xiao, S.; Gong, L.; Li, H.; Zhou, K.; Zhang, L.; Tu, Q.; Zhu, Y.Z.; Zhang, Y. Deciphering the nitrogen fixation gene cluster in vibrio natriegens: A study on optimized expression and application of nitrogenase. J. Agric. Food. Chem. 2024, 72, 12618–12629. [Google Scholar] [CrossRef]
- Smanski, M.J.; Bhatia, S.; Zhao, D.; Park, Y.; Woodruff, L.B.A.; Giannoukos, G.; Ciulla, D.; Busby, M.; Calderon, J.; Nicol, R.; et al. Functional optimization of gene clusters by combinatorial design and assembly. Nat. Biotechnol. 2014, 32, 1241–1249. [Google Scholar] [CrossRef]
- Addison, H.; Glatter, T.; Georg, K.A.H.; Rebelein Johannes, G. Two distinct ferredoxins are essential for nitrogen fixation by the iron nitrogenase in Rhodobacter capsulatus. mBio 2024, 15, e03314-23. [Google Scholar] [CrossRef]
- Ito, Y.; Yoshidome, D.; Hidaka, M.; Araki, Y.; Ito, K.; Kosono, S.; Nishiyama, M. Improvement of the nitrogenase activity in Escherichia coli that expresses the nitrogen fixation-related genes from Azotobacter vinelandii. Biochem. Biophys. Res. Commun. 2024, 728, 150345. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Gao, S.; Fu, J.; Li, R. Heterologous Expression of the Nitrogen-Fixing Gene Cluster from Paenibacillus polymyxa in Bacillus subtilis. Microorganisms 2025, 13, 1320. https://doi.org/10.3390/microorganisms13061320
Wang X, Gao S, Fu J, Li R. Heterologous Expression of the Nitrogen-Fixing Gene Cluster from Paenibacillus polymyxa in Bacillus subtilis. Microorganisms. 2025; 13(6):1320. https://doi.org/10.3390/microorganisms13061320
Chicago/Turabian StyleWang, Xiuling, Shiqing Gao, Jun Fu, and Ruijuan Li. 2025. "Heterologous Expression of the Nitrogen-Fixing Gene Cluster from Paenibacillus polymyxa in Bacillus subtilis" Microorganisms 13, no. 6: 1320. https://doi.org/10.3390/microorganisms13061320
APA StyleWang, X., Gao, S., Fu, J., & Li, R. (2025). Heterologous Expression of the Nitrogen-Fixing Gene Cluster from Paenibacillus polymyxa in Bacillus subtilis. Microorganisms, 13(6), 1320. https://doi.org/10.3390/microorganisms13061320