
Host–Microbiota Interactions in the Pathogenesis of Porcine Fetal Mummification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
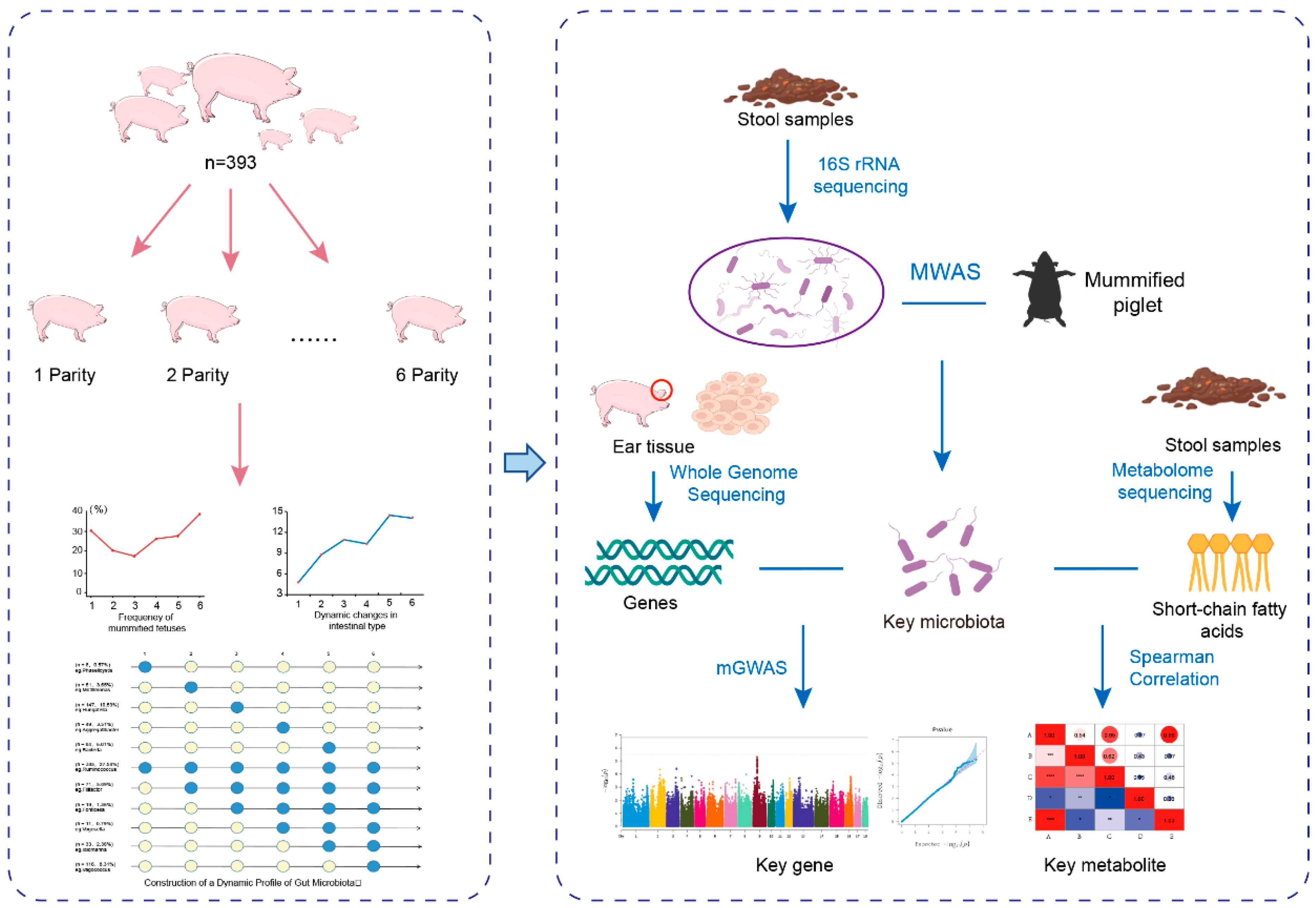
2. Materials and Methods
2.1. Animals and Phenotyping
2.2. Sample Collection
2.3. DNA Extraction and Polymerase Chain Reaction (PCR) Amplification
2.4. 16S rRNA Gene Sequence Assembly and Clustering
2.5. Bioinformatics and Statistical Analysis
2.6. Genotype Data Acquisition and Quality Control
2.7. Construction of a Map of Intestinal Microbes and Dynamic Changes in Mummium-Fetal Time
2.8. MWAS Analysis
2.9. mGWAS Analysis
2.10. Metabolomics Sequencing Analysis
3. Results
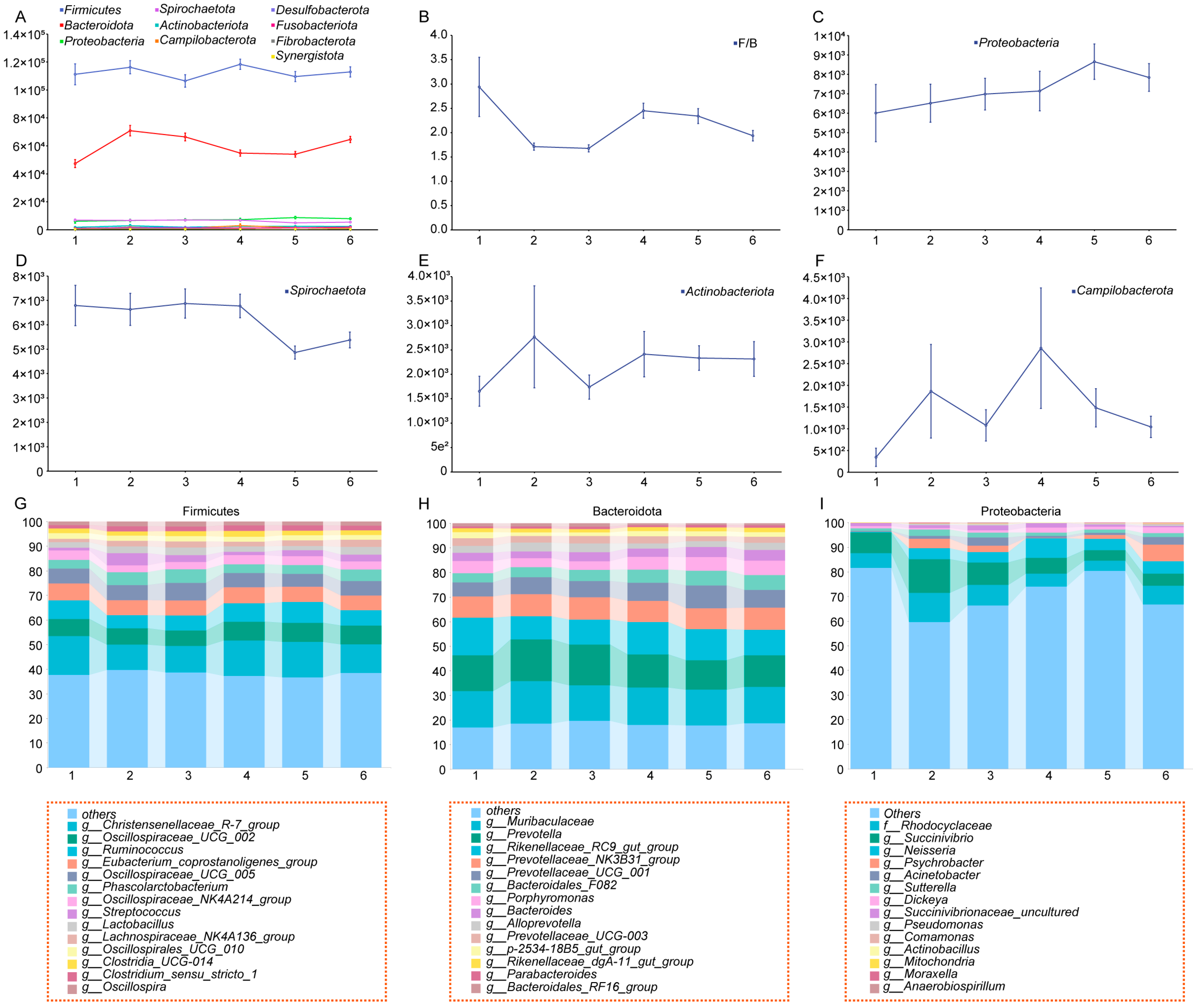
3.1. Frequency Analysis of Mummified Fetuses in Yorkshire Sows Across Different Parities and Dynamic Changes in Gut Microbiota Diversity
3.2. Construction of a Dynamic Profile of Gut Microbiota
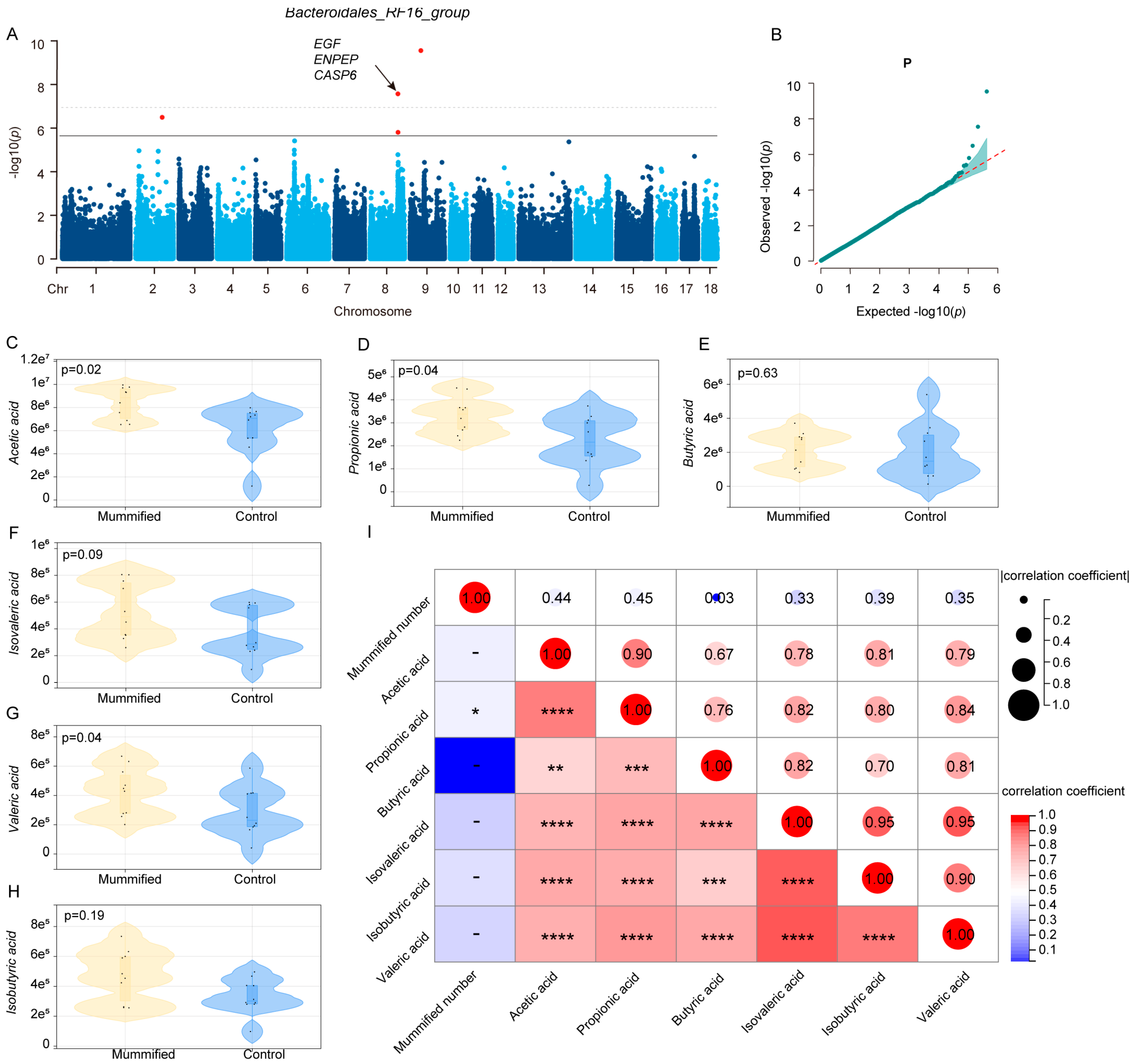
3.3. Identification of Microbiota Markers Significantly Associated with MUM Through Multi-Model MWAS
3.4. Association Between Host Genetics and the Key Microbiota Related to MUM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martin, F.P.; Wang, Y.; Sprenger, N.; Yap, I.K.; Lundstedt, T.; Lek, P.; Rezzi, S.; Ramadan, Z.; van Bladeren, P.; Fay, L.B.; et al. Probiotic modulation of symbi-otic gut microbial-host metabolic interactions in a humanized microbiome mouse model. Mol. Syst. Biol. 2008, 4, 157. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hu, J.; Ma, L.; Nie, Y.; Chen, J.; Zheng, W.; Wang, X.; Xie, C.; Zheng, Z.; Wang, Z.; Yang, T.; et al. A Microbiota-Derived Bacteriocin Targets the Host to Confer Diarrhea Resistance in Early-Weaned Piglets. Cell Host Microbe 2018, 24, 817–832.e8. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Li, H.; Gao, X.; Wang, L. Genome-wide association study of three litter traits in Yorkshire pigs. Anim. Genet. 2022, 53, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Mekonnen, K.T.; Lee, D.H.; Cho, Y.G.; Son, A.Y.; Seo, K.S. Genome-Wide Association Studies and Runs of Homozygosity Reveals Genetic Markers Associated with Reproductive Performance in Korean Duroc, Landrace, and Yorkshire Breeds. Genes 2024, 15, 1422. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tantasuparuk, W.; Lundeheim, N.; Dalin, A.M.; Kunavongkrit, A.; Einarsson, S. Reproductive performance of purebred landrace and Yorkshire sows in Thailand with special reference to seasonal influence and parity number. Theriogenology 2000, 54, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Wang, X.; Wang, J.; Zhao, J. Life-long dynamics of the swine gut microbiome and their implications in probiotics development and food safety. Gut Microbes 2020, 11, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Deng, F.; Li, Y.; Zhao, J. Identification of gut microbiome signatures associated with longevity provides a promising modulation target for healthy aging. Gut Microbes 2019, 10, 210–215. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wei, X.; Tsai, T.; Knapp, J.; Bottoms, K.; Deng, F.; Story, R.; Maxwell, C.; Zhao, J. ZnO Modulates Swine Gut Microbiota and Improves Growth Performance of Nursery Pigs When Combined with Peptide Cocktail. Microorganisms 2020, 8, 146. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guo, X.; Su, G.; Christensen, O.F.; Janss, L.; Lund, M.S. Genome-wide association analyses using a Bayesian approach for litter size and piglet mortality in Danish Landrace and Yorkshire pigs. BMC Genom. 2016, 17, 468. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, Z.; Ye, S.; Teng, J.; Diao, S.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J.; Zhang, Z. Genome-wide association studies for the number of animals born alive and dead in duroc pigs. Theriogenology 2019, 139, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Derks, M.F.L.; Megens, H.J.; Bosse, M.; Lopes, M.S.; Harlizius, B.; Groenen, M.A.M. A systematic survey to identify lethal recessive variation in highly managed pig populations. BMC Genom. 2017, 18, 858. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dron, N.; Hernández-Jover, M.; Doyle, R.E.; Holyoake, P.K. Investigating risk factors and possible infectious aetiologies of mummified fetuses on a large piggery in Australia. Aust. Vet. J. 2014, 92, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, S.; Krusche, P.; Dolzhenko, E.; Sherman, R.M.; Petrovski, R.; Schlesinger, F.; Kirsche, M.; Bentley, D.R.; Schatz, M.C.; Sedlazeck, F.J.; et al. Paragraph: A graph-based structural variant genotyper for short-read sequence data. Genome Biol. 2019, 20, 291. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wen, C.; Yan, W.; Sun, C.; Ji, C.; Zhou, Q.; Zhang, D.; Zheng, J.; Yang, N. The gut microbiota is largely independent of host genetics in regulating fat deposition in chickens. ISME J. 2019, 13, 1422–1436. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gao, X.; Starmer, J.; Martin, E.R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 2008, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giron, M.; Thomas, M.; Jarzaguet, M.; Mayeur, C.; Ferrere, G.; Noordine, M.L.; Bornes, S.; Dardevet, D.; Chassard, C.; Savary-Auzeloux, I. Lacticaseibacillus casei CNCM I-5663 supplementation maintained muscle mass in a model of frail rodents. Front. Nutr. 2022, 9, 928798. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bakoev, S.; Getmantseva, L.; Bakoev, F.; Kolosova, M.; Gabova, V.; Kolosov, A.; Kostyunina, O. Survey of SNPs Associated with Total Number Born and Total Number Born Alive in Pig. Genes 2020, 11, 491. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yan, W.; Sun, C.; Yuan, J.; Yang, N. Gut metagenomic analysis reveals prominent roles of Lactobacillus and cecal microbiota in chicken feed efficiency. Sci. Rep. 2017, 7, 45308. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shah, T.M.; Patel, J.G.; Gohil, T.P.; Blake, D.P.; Joshi, C.G. Host transcriptome and microbiome interaction modulates physiology of full-sibs broilers with divergent feed conversion ratio. NPJ Biofilms Microbiomes 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Grice, E.A.; Segre, J.A. The human microbiome: Our second genome. Annu. Rev. Genom. Hum. Genet. 2012, 13, 151–170. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Turnbaugh, P.; Ley, R.; Mahowald, M.; Magrini, V.; Mardis, E.; Gordon, J.J.N. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Rubino, F.; Carberry, C.; Waters, S.M.; Kenny, D.; McCabe, M.S.; Creevey, C.J. Divergent functional isoforms drive niche specialisation for nutrient acquisition and use in rumen microbiome. ISME J. 2017, 11, 932–944. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, H.; Ji, Y.; Yin, C.; Deng, M.; Tang, T.; Deng, B.; Ren, W.; Deng, J.; Yin, Y.; Tan, C. Differential Analysis of Gut Microbiota Correlated With Oxidative Stress in Sows With High or Low Litter Performance During Lactation. Front. Microbiol. 2018, 9, 1665. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carney-Hinkle, E.E.; Tran, H.; Bundy, J.W.; Moreno, R.; Miller, P.S.; Burkey, T.E. Effect of dam parity on litter performance, transfer of passive immunity, and progeny microbial ecology. J. Anim. Sci. 2013, 91, 2885–2893. [Google Scholar] [CrossRef] [PubMed]
- Kemme, P.A.; Radcliffe, J.S.; Jongbloed, A.W.; Mroz, Z. The effects of sow parity on digestibility of proximate components and minerals during lactation as influenced by diet and microbial phytase supplementation. J. Anim. Sci. 1997, 75, 2147–2153. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Christensen, L.; Roager, H.M.; Astrup, A.; Hjorth, M.F. Microbial enterotypes in personalized nutrition and obesity management. Am. J. Clin. Nutr. 2018, 108, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Spychala, M.S.; Venna, V.R.; Jandzinski, M.; Doran, S.J.; Durgan, D.J.; Ganesh, B.P.; Ajami, N.J.; Putluri, N.; Graf, J.; Bryan, R.M.; et al. Age-related changes in the gut microbiota influence systemic inflammation and stroke outcome. Ann. Neurol. 2018, 84, 23–36. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ahmed, K.; Choi, H.N.; Cho, S.R.; Yim, J.E. Association of Firmicutes/Bacteroidetes Ratio with Body Mass Index in Korean Type 2 Diabetes Mellitus Patients. Metabolites 2024, 14, 518. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lin, Y.; Zhou, B.; Zhu, W. Pathogenic Escherichia coli-Specific Bacteriophages and Polyvalent Bacteriophages in Piglet Guts with Increasing Coliphage Numbers after Weaning. Appl. Environ. Microbiol. 2021, 87, e0096621. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ji, X.; Liang, B.; Sun, Y.; Zhu, L.; Zhou, B.; Guo, X.; Liu, J. An Extended-Spectrum Beta-Lactamase-Producing Hybrid Shiga-Toxigenic and Enterotoxigenic Escherichia coli Strain Isolated from a Piglet with Diarrheal Disease in Northeast China. Foodborne Pathog. Dis. 2020, 17, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, Y.; Chen, X.; Fang, C.; Zhao, L.; Chen, F. The Maturing Development of Gut Microbiota in Commercial Piglets during the Weaning Transition. Front. Microbiol. 2017, 8, 1688. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- He, J.; Zheng, W.; Tao, C.; Guo, H.; Xue, Y.; Zhao, R.; Yao, W. Heat stress during late gestation disrupts maternal microbial transmission with altered offspring’s gut microbial colonization and serum metabolites in a pig model. Environ. Pollut. 2020, 266 Pt 3, 115111. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Li, H.; Li, D.; Peng, J.; Zhang, X.; Yang, W. The Gut Microbiota of Pregnant Rats Alleviates Fetal Growth Restriction by Inhibiting the TLR9/MyD88 Pathway. J. Microbiol. Biotechnol. 2023, 33, 1213–1227. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zheng, J.; Li, S.; He, J.; Liu, H.; Huang, Y.; Jiang, X.; Zhao, X.; Li, J.; Feng, B.; Che, L.; et al. A Gestational Pectin Diet Could Improve the Health of Multiparous Sows by Modulating the Gut Microbiota and Cytokine Level during Late Pregnancy. Animals 2024, 14, 1559. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Pregnancy-Specific β1-Glycoproteins: Combined Biomarker Roles, Structure/Function Relationships and Implications for Drug Design. Curr. Med. Chem. 2017, 24, 245–267. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37 (Suppl. S4), S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Shirakata, Y.; Kimura, R.; Nanba, D.; Iwamoto, R.; Tokumaru, S.; Morimoto, C.; Yokota, K.; Nakamura, M.; Sayama, K.; Mekada, E.; et al. Heparin-binding EGF-like growth factor accelerates keratinocyte migration and skin wound healing. J. Cell Sci. 2005, 118 Pt 11, 2363–2370. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, A.C.H.; Wei, X.; Fong, S.W.; Yeung, W.S.B.; Lee, Y.L. Uncovering the role of TET2-mediated ENPEP activation in trophoblast cell fate determination. Cell. Mol. Life Sci. 2024, 81, 270. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aung, M.; Konoshita, T.; Moodley, J.; Naicker, T.; Connolly, C.; Khaliq, O.P.; Gathiram, P. Aminopeptidase A (ENPEP) gene polymorphisms and preeclampsia: Descriptive analysis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 258, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.; Yoo, I.; Han, J.; Kim, M.; Lee, S.; Cheon, Y.; Hong, M.; Jeon, B.-Y.; Ka, H. Expression of Caspases in the Pig Endome-trium Throughout the Estrous Cycle and at the Maternal-Conceptus Interface During Pregnancy and Regulation by Steroid Hormones and Cytokines. Front. Vet. Sci. 2021, 8, 641916. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cavalcanti, M.C.; Steilmann, C.; Failing, K.; Bergmann, M.; Kliesch, S.; Weidner, W.; Steger, K. Apoptotic gene expression in potentially fertile and subfertile men. Mol. Hum. Reprod. 2011, 17, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, M.; Ke, S.; Huang, X.; Fang, S.; He, M.; Fu, H.; Chen, C.; Huang, L. Gut and Vagina Microbiota Associated with Estrus Return of Weaning Sows and Its Correlation with the Changes in Serum Metabolites. Front. Microbiol. 2021, 12, 690091. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ma, C.; Gao, Q.; Zhang, W.; Azad, M.A.K.; Kong, X. Alterations in the Blood Parameters and Fecal Microbiota and Metabolites during Pregnant and Lactating Stages in Bama Mini Pigs as a Model. Mediat. Inflamm. 2020, 2020, 8829072. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cooke, P.S.; Ekman, G.C.; Kaur, J.; Davila, J.; Bagchi, I.C.; Clark, S.G.; Dziuk, P.J.; Hayashi, K.; Bartol, F.F. Brief exposure to progesterone during a critical neonatal window prevents uterine gland formation in mice. Biol. Reprod. 2012, 86, 63. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sam, Q.H.; Ling, H.; Yew, W.S.; Tan, Z.; Ravikumar, S.; Chang, M.W.; Chai, L.Y.A. The Divergent Immunomodulatory Effects of Short Chain Fatty Acids and Medium Chain Fatty Acids. Int. J. Mol. Sci. 2021, 22, 6453. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sakata, T. Pitfalls in short-chain fatty acid research: A methodological review. Anim. Sci. J. 2019, 90, 3–13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, W.; Sun, M.; Chen, F.; Cao, A.T.; Liu, H.; Zhao, Y.; Huang, X.; Xiao, Y.; Yao, S.; Zhao, Q.; et al. Microbiota metabolite short-chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol. 2017, 10, 946–956. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Zhang, L.; Liu, Z.; Guo, A.; Yang, G.; Yu, T. Host–Microbiota Interactions in the Pathogenesis of Porcine Fetal Mummification. Microorganisms 2025, 13, 1052. https://doi.org/10.3390/microorganisms13051052
Wang M, Zhang L, Liu Z, Guo A, Yang G, Yu T. Host–Microbiota Interactions in the Pathogenesis of Porcine Fetal Mummification. Microorganisms. 2025; 13(5):1052. https://doi.org/10.3390/microorganisms13051052
Chicago/Turabian StyleWang, Mingyu, Lin Zhang, Zhe Liu, Ao Guo, Gongshe Yang, and Taiyong Yu. 2025. "Host–Microbiota Interactions in the Pathogenesis of Porcine Fetal Mummification" Microorganisms 13, no. 5: 1052. https://doi.org/10.3390/microorganisms13051052
APA StyleWang, M., Zhang, L., Liu, Z., Guo, A., Yang, G., & Yu, T. (2025). Host–Microbiota Interactions in the Pathogenesis of Porcine Fetal Mummification. Microorganisms, 13(5), 1052. https://doi.org/10.3390/microorganisms13051052