
Comparative Genomic and Transcriptomic Analysis Provides New Insights into the Aflatoxin B1 Biodegradability by Kocuria rosea from Deep Sea


Abstract
1. Introduction
2. Materials and Methods
2.1. Comparative Genomic Analysis
2.2. Strain and Chemical Preparation
2.3. Total RNA Extraction and Quality Detection
2.4. RNA Library Construction, Library Enrichment, and Sequencing
2.5. Bioinformatics Analysis of Transcriptome Sequencing Data
3. Results and Discussion
3.1. Genome Characteristics of Genus Kocuria Strains
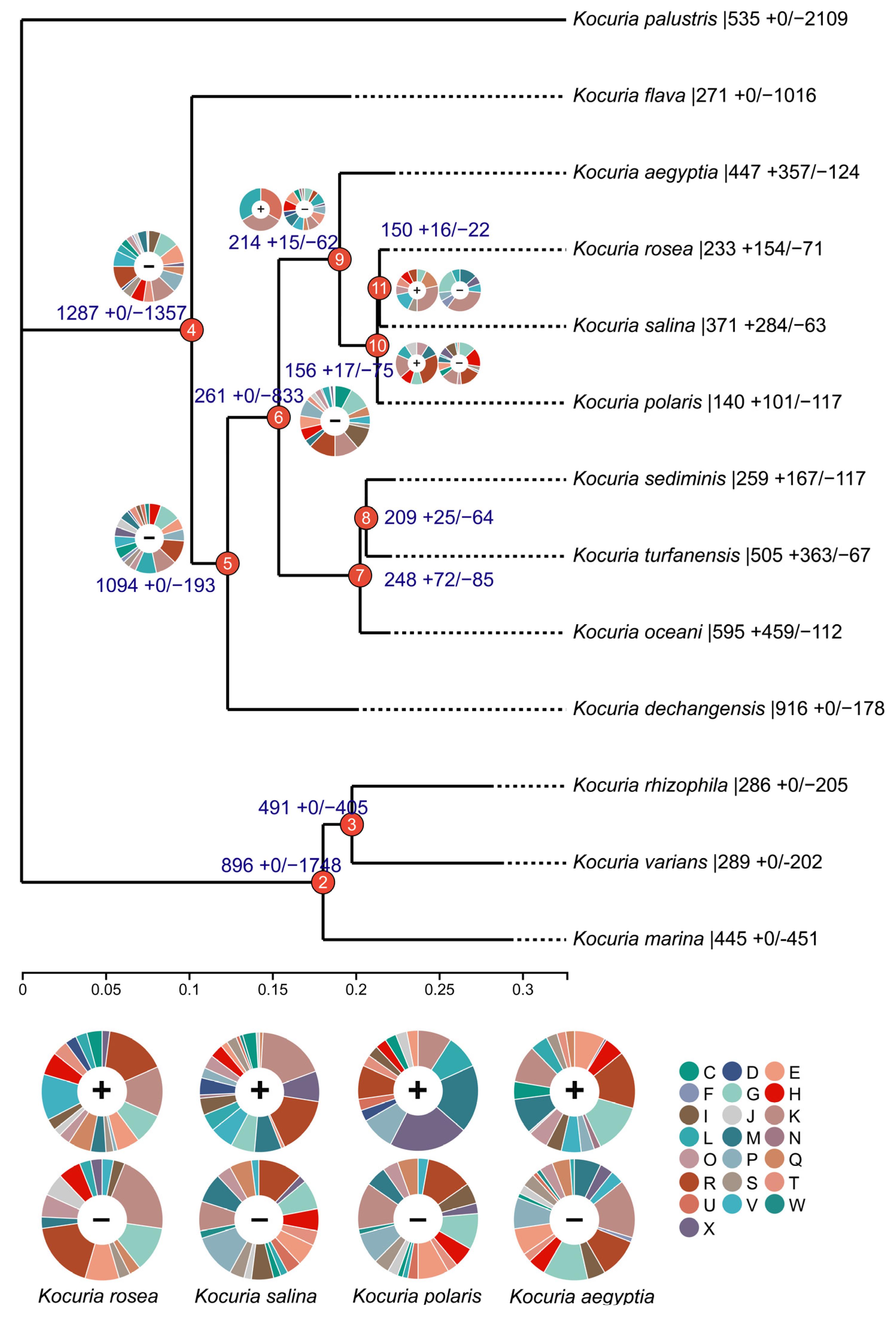
3.2. Phylogenetic Analysis of Genus Kocuria
3.3. Pan-Genome and Core Genome Analysis of the Genus Kocuria Strains
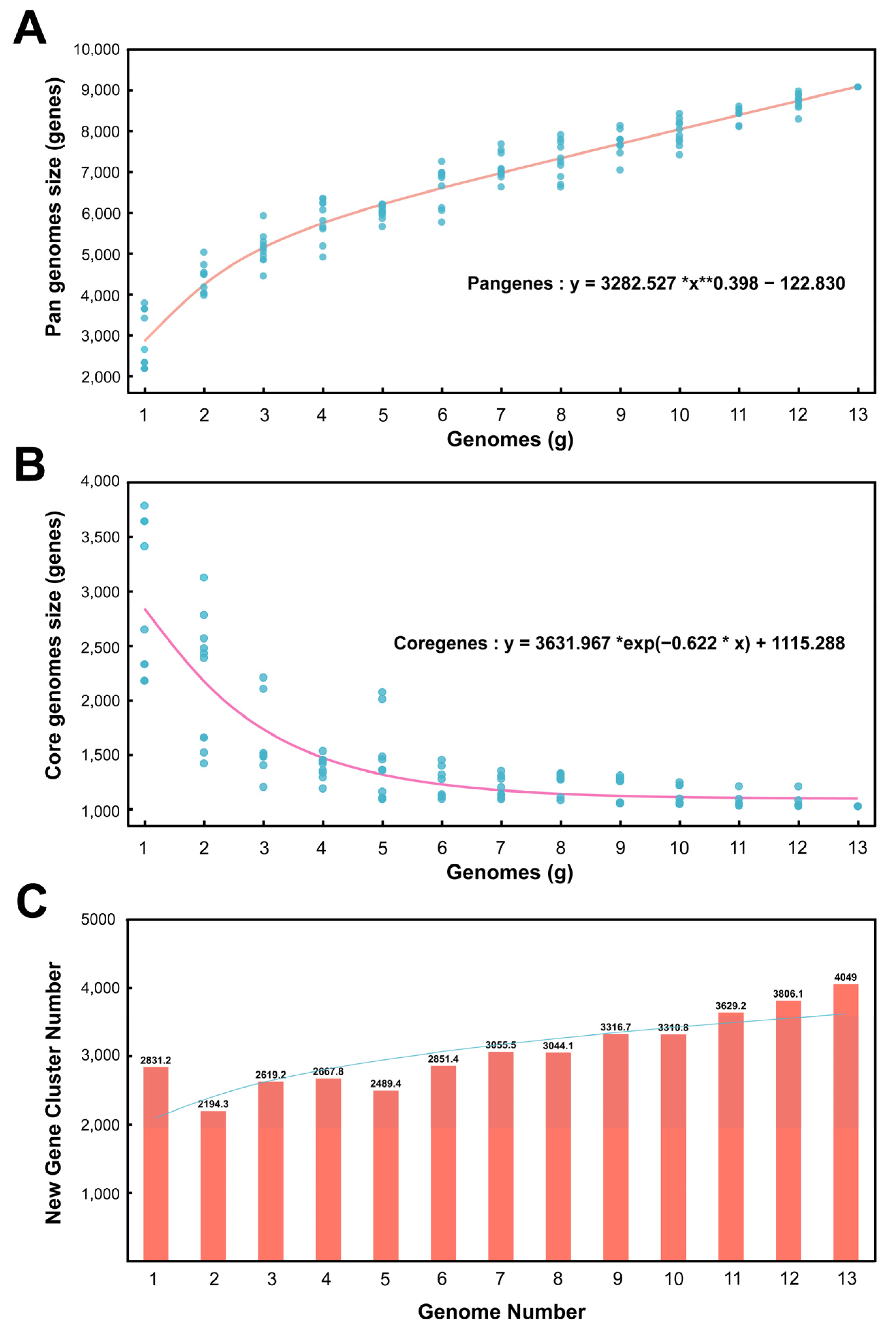
3.3.1. Pan-Genomic Quantitative Analysis
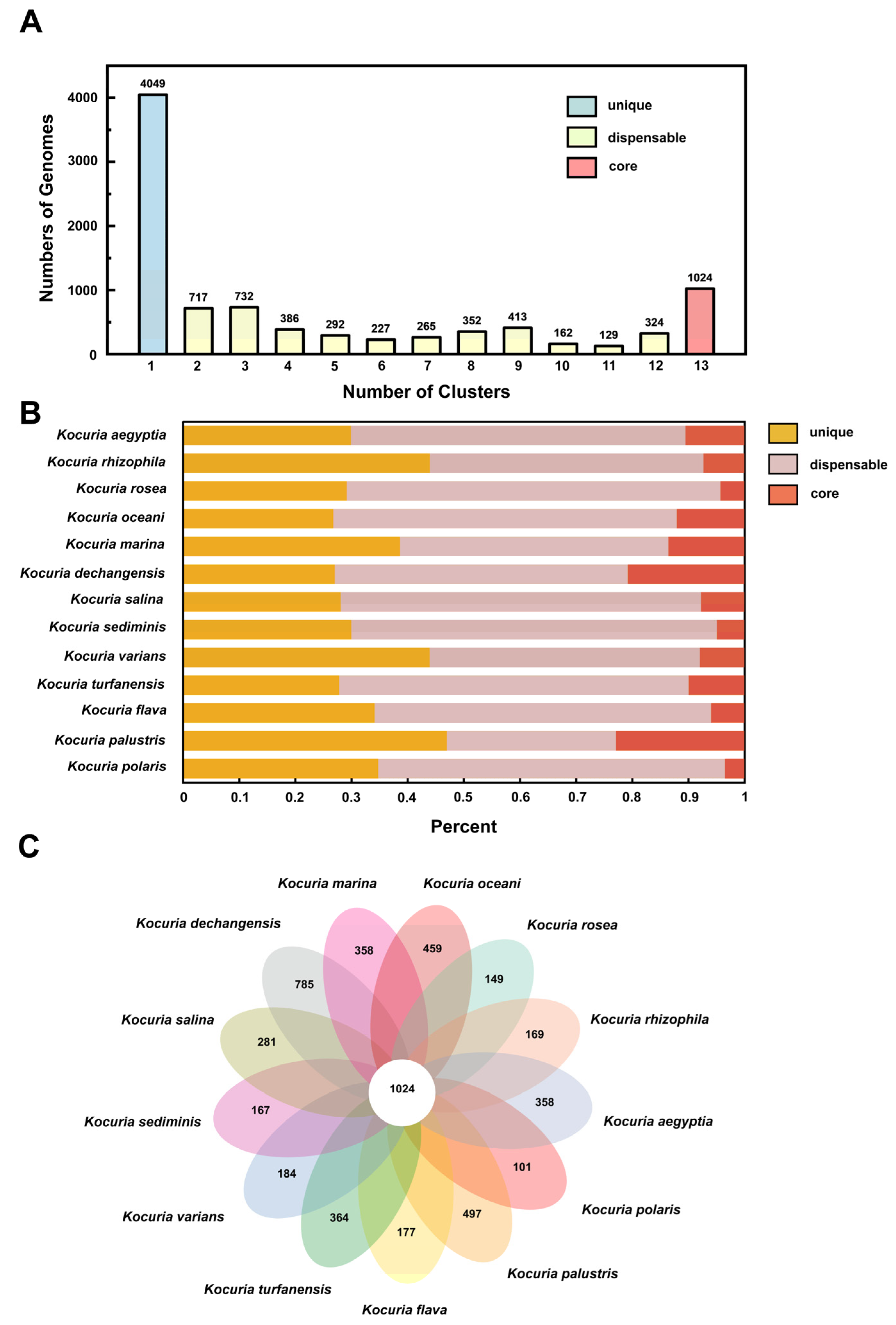
3.3.2. Classification and Distribution of Homologous Genes
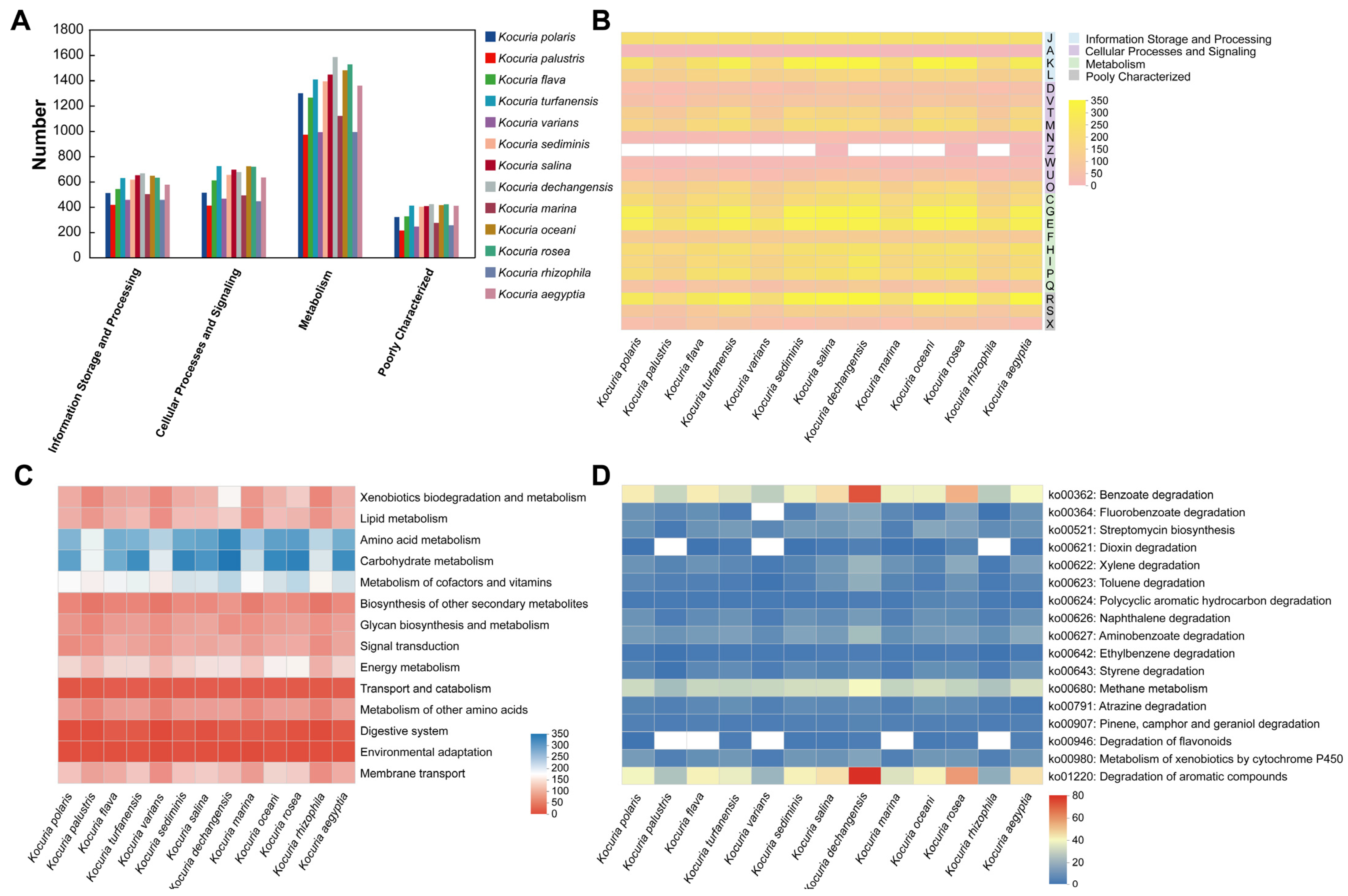
3.4. Comparative Gene Function Analysis of 13 Strains in the Genus Kocuria
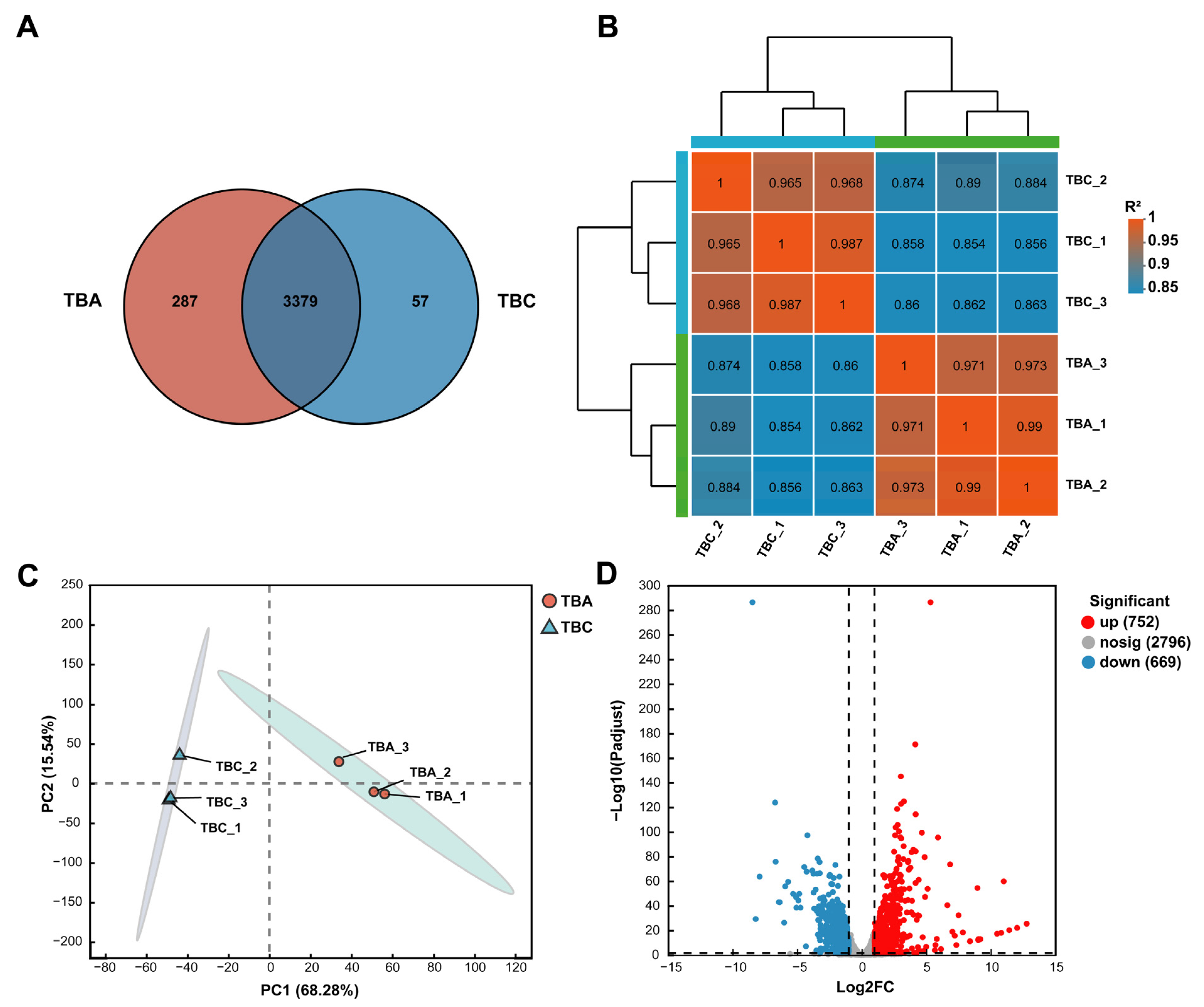
3.5. Analysis of Gene Expression Correlation and Gene Expression Difference
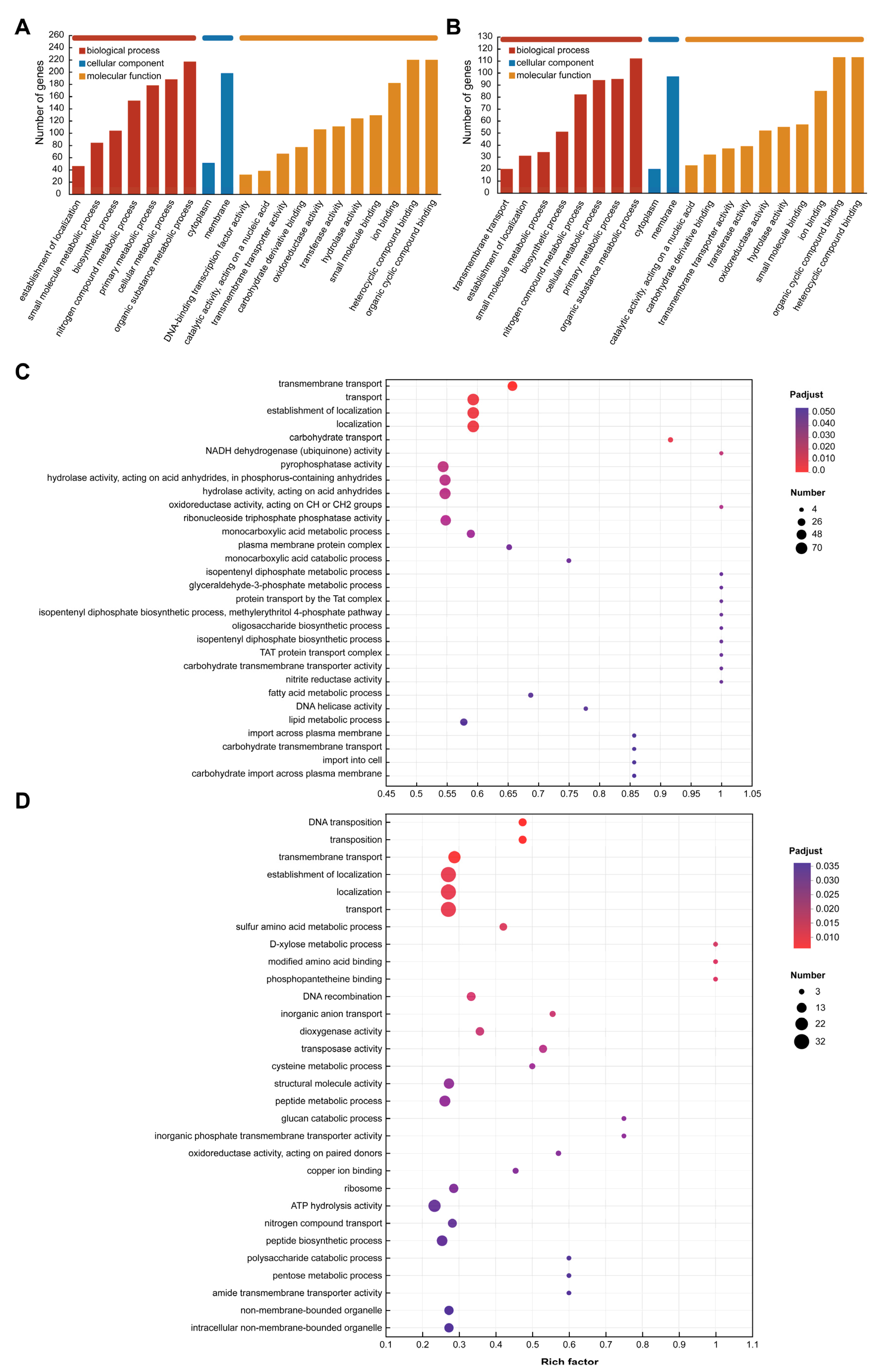
3.6. Differently Expressed Gene Function Analysis
3.7. Analysis of AFB1 Degradation-Related Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, J.; Qin, X.J.; Guo, Y.P.; Zhang, Q.Q.; Ma, Q.G.; Ji, C.; Zhao, L.H. Enzymatic degradation of deoxynivalenol by a novel bacterium, Pelagibacterium halotolerans ANSP101. Food Chem. Toxicol. 2020, 140, 8. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 2007, 5, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Bosch, R.; García-Valdés, E.; Moore, E.R.B. Complete nucleotide sequence and evolutionary significance of a chromosomally encoded naphthalene-degradation lower pathway from Pseudomonas stutzeri AN10. Gene 2000, 245, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.H.; Choi, C.W.; Lee, S.Y.; Lee, Y.G.; Kwon, J.; Leem, S.H.; Chung, Y.H.; Kahng, H.Y.; Kim, S.J.; Kwon, K.K.; et al. Proteomic Characterization of Plasmid pLA1 for Biodegradation of Polycyclic Aromatic Hydrocarbons in the Marine Bacterium, Novosphingobium pentaromativorans US6-1. PLoS ONE 2014, 9, 11. [Google Scholar] [CrossRef]
- Cao, J.; Lai, Q.; Yuan, J.; Shao, Z. Genomic and metabolic analysis of fluoranthene degradation pathway in Celeribacter indicus P73T. Sci. Rep. 2015, 5, 7741. [Google Scholar]
- Hu, P.; Dubinsky, E.A.; Probst, A.J.; Wang, J.; Sieber, C.M.K.; Tom, L.M.; Gardinali, P.R.; Banfield, J.F.; Atlas, R.M.; Andersen, G.L. Simulation of Deepwater Horizon oil plume reveals substrate specialization within a complex community of hydrocarbon degraders. Proc. Natl. Acad. Sci. USA 2017, 114, 7432–7437. [Google Scholar] [CrossRef]
- Sarkhel, R.; Sengupta, S.; Das, P.; Bhowal, A. Comparative biodegradation study of polymer from plastic bottle waste using novel isolated bacteria and fungi from marine source. J. Polym. Res. 2020, 27, 8. [Google Scholar] [CrossRef]
- Paço, A.; Duarte, K.; da Costa, J.P.; Santos, P.S.M.; Pereira, R.; Pereira, M.E.; Freitas, A.C.; Duarte, A.C.; Rocha-Santos, T.A.P. Biodegradation of polyethylene microplastics by the marine fungus Zalerion maritimum. Sci. Total Environ. 2017, 586, 10–15. [Google Scholar] [CrossRef]
- Varjani, S.J. Microbial degradation of petroleum hydrocarbons. Bioresour. Technol. 2017, 223, 277–286. [Google Scholar] [CrossRef]
- Tao, L.Y.; Zhang, J.Y.; Liang, Y.J.; Chen, L.M.; Zheng, L.S.; Wang, F.; Mi, Y.J.; She, Z.G.; To, K.K.W.; Lin, Y.C.; et al. Anticancer Effect and Structure-Activity Analysis of Marine Products Isolated from Metabolites of Mangrove Fungi in the South China Sea. Mar. Drugs 2010, 8, 1094–1105. [Google Scholar] [CrossRef]
- Garo, E.; Starks, C.M.; Jensen, P.R.; Fenical, W.; Lobkovsky, E.; Clardy, J. Trichodermamides A and B, cytotoxic modified Dipeptides from the marine-derived fungus Trichoderma virens. J. Nat. Prod. 2003, 66, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gu, Q.Q.; Zhu, W.M.; Cui, C.B.; Fan, G.T. Trichodermamide A and aspergillazine A, two cytotoxic modified dipeptides from a marine-derived fungus Spicaria elegans. Arch. Pharm. Res. 2005, 28, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, Z.Y.; Sun, K.L.; Zhu, W.M. Effects of High Salt Stress on Secondary Metabolite Production in the Marine-Derived Fungus Spicaria elegans. Mar. Drugs 2011, 9, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Hokari, R.; Ishiyama, A.; Iwatsuki, M.; Higo, M.; Nonaka, K.; Nagano, Y.; Takahashi, Y.; Omura, S.; Nakashima, T. Hatsusamides A and B: Two New Metabolites Produced by the Deep-Sea-Derived Fungal Strain Penicillium steckii FKJ-0213. Mar. Drugs 2020, 18, 513. [Google Scholar] [CrossRef]
- Huang, Z.J.; Yang, J.X.; Cai, X.L.; She, Z.G.; Lin, Y.C. A new furanocoumarin from the mangrove endophytic fungus Penicillium sp (ZH16). Nat. Prod. Res. 2012, 26, 1291–1295. [Google Scholar] [CrossRef]
- Tan, N.; Tao, Y.W.; Pan, J.H.; Wang, S.Y.; Xu, F.; She, Z.G.; Lin, Y.C.; Jones, E.B.G. Isolation, structure elucidation, and mutagenicity of four alternariol derivatives produced by the mangrove endophytic fungus No. 2240. Chem. Nat. Compd. 2008, 44, 296–300. [Google Scholar] [CrossRef]
- Lin, A.Q.; Du, L.; Fang, Y.C.; Wang, F.Z.; Zhu, T.J.; Gu, Q.Q.; Zhu, W.M. iso-α-Cyclopiazonic acid, a new natural product isolated from the marine-derived fungus Aspergillus flavus C-F-3. Chem. Nat. Compd. 2009, 45, 677–680. [Google Scholar] [CrossRef]
- Wang, H.; Lu, Z.Y.; Qu, H.J.; Liu, P.P.; Miao, C.D.; Zhu, T.H.; Li, J.; Hong, K.; Zhu, W.M. Antimicrobial aflatoxins from the marine-derived fungus Aspergillus flavus 092008. Arch. Pharm. Res. 2012, 35, 1387–1392. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Samson, R.A. Emericella venezuelensis, a new species with stellate ascospores producing sterigmatocystin and aflatoxin B1. Syst. Appl. Microbiol. 2004, 27, 672–680. [Google Scholar] [CrossRef]
- Zijden, A.S.M.V.D.; Koelensmid, W.A.A.B.; Boldingh, J. Aspergillus flavus and Turkey X Disease: Isolation in Crystalline Form of a Toxin responsible for Turkey X Disease. Nature 1962, 195, 1060–1062. [Google Scholar]
- Di Stefano, V.; Pitonzo, R.; Cicero, N.; D’Oca, M.C. Mycotoxin contamination of animal feedingstuff: Detoxification by gamma-irradiation and reduction of aflatoxins and ochratoxin A concentrations. Food Addit. Contam. Part A Chem. 2014, 31, 2034–2039. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.T.; Hirooka, E.Y.; Silva, P.; Bracarense, A.; Flaiban, K.; Akagi, C.Y.; Kawamura, O.; da Costa, M.C.; Itano, E.N. Impact of a Single Oral Acute Dose of Aflatoxin B1 on Liver Function/Cytokines and the Lymphoproliferative Response in C57Bl/6 Mice. Toxins 2017, 9, 374. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, M.T.; Netzel, G. Food Safety and Natural Toxins. Toxins 2020, 12, 236. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Cotty, P.J. Aflatoxin contamination of dried red chilies: Contrasts between the United States and Nigeria, two markets differing in regulation enforcement. Food Control 2017, 80, 374–379. [Google Scholar] [CrossRef]
- Rahaie, S.; Emam-Djomeh, Z.; Razavi, S.H.; Mazaheri, M. Evaluation of aflatoxin decontaminating by two strains of Saccharomyces cerevisiae and Lactobacillus rhamnosus strain GG in pistachio nuts. Int. J. Food Sci. Technol. 2012, 47, 1647–1653. [Google Scholar] [CrossRef]
- Branà, M.T.; Cimmarusti, M.T.; Haidukowski, M.; Logrieco, A.F.; Altomare, C. Bioremediation of aflatoxin B1-contaminated maize by king oyster mushroom (Pleurotus eryngii). PLoS ONE 2017, 12, 14. [Google Scholar] [CrossRef]
- Das, A.; Bhattacharya, S.; Palaniswamy, M.; Angayarkanni, J. Biodegradation of aflatoxin B1 in contaminated rice straw by Pleurotus ostreatus MTCC 142 and Pleurotus ostreatus GHBBF10 in the presence of metal salts and surfactants. World J. Microbiol. Biotechnol. 2014, 30, 2315–2324. [Google Scholar] [CrossRef]
- Wang, L.L.; Wu, J.; Liu, Z.W.; Shi, Y.T.; Liu, J.Q.; Xu, X.F.; Hao, S.X.; Mu, P.Q.; Deng, F.R.; Deng, Y.Q. Aflatoxin B1 Degradation and Detoxification by Escherichia coli CG1061 Isolated from Chicken Cecum. Front. Pharmacol. 2019, 9, 9. [Google Scholar] [CrossRef]
- Alberts, J.F.; Gelderblom, W.C.A.; Botha, A.; van Zyl, W.H. Degradation of aflatoxin B1 by fungal laccase enzymes. Int. J. Food Microbiol. 2009, 135, 47–52. [Google Scholar] [CrossRef]
- Wang, J.Q.; Ogata, M.; Hirai, H.; Kawagishi, H. Detoxification of aflatoxin B1 by manganese peroxidase from the white-rot fungus Phanerochaete sordida YK-624. FEMS Microbiol. Lett. 2011, 314, 164–169. [Google Scholar] [CrossRef]
- Taylor, M.C.; Jackson, C.J.; Tattersall, D.B.; French, N.; Peat, T.S.; Newman, J.; Briggs, L.J.; Lapalikar, G.V.; Campbell, P.M.; Scott, C.; et al. Identification and characterization of two families of F420H2-dependent reductases from Mycobacteria that catalyse aflatoxin degradation. Mol. Microbiol. 2010, 78, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Z.; Lu, F.P.; Jiang, H.L.; Tan, C.P.; Yao, D.S.; Xie, C.F.; Liu, D.L. The furofuran-ring selectivity, hydrogen peroxide-production and low Km value are the three elements for highly effective detoxification of aflatoxin oxidase. Food Chem. Toxicol. 2015, 76, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Nan, J.; Chen, Q.Q.; Zhou, Y.; Gao, X.J.; Li, Y.X. Exploration of Aflatoxin B1 Degradation Products via Kocuria rosea: Structure Elucidation and Toxicity Analysis. Appl. Sci. 2024, 14, 11024. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, X.Y.; Chen, H.Y.; Huang, W.N.; Jiang, H.N.; Wang, C.L.; Xiao, Z.; Zhang, Y.Y.; Xu, J.L. Isolation and Aflatoxin B1-Degradation Characteristics of a Microbacterium proteolyticum B204 Strain from Bovine Faeces. Toxins 2022, 14, 525. [Google Scholar] [CrossRef]
- Shu, X.; Wang, Y.T.; Zhou, Q.; Li, M.H.; Hu, H.; Ma, Y.H.; Chen, X.; Ni, J.; Zhao, W.W.; Huang, S.W.; et al. Biological Degradation of Aflatoxin B1 by Cell-Free Extracts of Bacillus velezensis DY3108 with Broad PH Stability and Excellent Thermostability. Toxins 2018, 10, 330. [Google Scholar] [CrossRef]
- Zhang, W.; Xue, B.B.; Li, M.M.; Mu, Y.; Chen, Z.H.; Li, J.P.; Shan, A.S. Screening a Strain of Aspergillus niger and Optimization of Fermentation Conditions for Degradation of Aflatoxin B1. Toxins 2014, 6, 3157–3172. [Google Scholar] [CrossRef]
- Hua, S.S.T.; Sarreal, S.B.L.; Chang, P.K.; Yu, J.J. Transcriptional Regulation of Aflatoxin Biosynthesis and Conidiation in Aspergillus flavus by Wickerhamomyces anomalus WRL-076 for Reduction of Aflatoxin Contamination. Toxins 2019, 11, 81. [Google Scholar] [CrossRef]
- El-Deeb, B.; Altalhi, A.; Khiralla, G.; Hassan, S.; Gherbawy, Y. Isolation and Characterization of Endophytic Bacilli Bacterium from Maize Grains Able to Detoxify Aflatoxin B1. Food Biotechnol. 2013, 27, 199–212. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Koch, C.; Gvozdiak, O.; Schumann, P. Taxonomic Dissection of the Genus Micrococcus: Kocuria Gen. Nov., Nesterenkonia Gen. Nov., Kytococcus Gen. Nov., Dermacoccus Gen. Nov. and Micrococcus Cohn 1872 Gen. Emend. Int. J. Syst. Bacteriol. 1995, 45, 682–692. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Frederiksen, W.; Garrity, G.M.; Grimont, P.A.D.; Kämpfer, P.; Maiden, M.C.J.; Nesme, X.; Rosselló-Mora, R.; Swings, J.; Trüper, H.G.; et al. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 2002, 52, 1043–1047. [Google Scholar] [CrossRef]
- Achal, V.; Pan, X.L.; Zhang, D.Y. Remediation of copper-contaminated soil by Kocuria flava CR1, based on microbially induced calcite precipitation. Ecol. Eng. 2011, 37, 1601–1605. [Google Scholar] [CrossRef]
- Callejón, S.; Sendra, R.; Ferrer, S.; Pardo, I. Ability of Kocuria varians LTH 1540 To Degrade Putrescine: Identification and Characterization of a Novel Amine Oxidase. J. Agric. Food Chem. 2015, 63, 4170–4178. [Google Scholar] [CrossRef] [PubMed]
- Haq, F.; Butt, M.; Ali, H.; Chaudhary, H.J. Biosorption of cadmium and chromium from water by endophytic Kocuria rhizophila: Equilibrium and kinetic studies. Desalin. Water Treat. 2016, 57, 19946–19958. [Google Scholar] [CrossRef]
- Hussain, A.; Amna; Kamran, M.A.; Javed, M.T.; Hayat, K.; Farooq, M.A.; Ali, N.; Ali, M.; Manghwar, H.; Jan, F.; et al. Individual and combinatorial application of Kocuria rhizophila and citric acid on phytoextraction of multi-metal contaminated soils by Glycine max L. Environ. Exp. Bot. 2019, 159, 23–33. [Google Scholar] [CrossRef]
- Vidal, L.; Christen, P.; Coello, M.N. Feather degradation by Kocuria rosea in submerged culture. World J. Microbiol. Biotechnol. 2000, 16, 551–554. [Google Scholar] [CrossRef]
- Bertsch, A.; Coello, N. A biotechnological process for treatment and recycling poultry feathers as a feed ingredient. Bioresour. Technol. 2005, 96, 1703–1708. [Google Scholar] [CrossRef]
- Parshetti, G.K.; Parshetti, S.; Kalyani, D.C.; Doong, R.A.; Govindwar, S.P. Industrial dye decolorizing lignin peroxidase from Kocuria rosea MTCC 1532. Ann. Microbiol. 2012, 62, 217–223. [Google Scholar] [CrossRef]
- Carretero-Paulet, L.; Chang, T.H.; Librado, P.; Ibarra-Laclette, E.; Herrera-Estrella, L.; Rozas, J.; Albert, V.A. Genome-Wide Analysis of Adaptive Molecular Evolution in the Carnivorous Plant Utricularia gibba. Genome Biol. Evol. 2015, 7, 444–456. [Google Scholar] [CrossRef]
- Zhong, C.F.; Han, M.Z.; Yu, S.J.; Yang, P.S.; Li, H.J.; Ning, K. Pan-genome analyses of 24 Shewanella strains re-emphasize the diversification of their functions yet evolutionary dynamics of metal-reducing pathway. Biotechnol. Biofuels 2018, 11, 13. [Google Scholar] [CrossRef]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- An, X.J.; Li, N.J.; Zhang, L.Z.; Xu, Z.H.; Zhang, S.L.; Zhang, Q.H. New insights into the typical nitrogen-containing heterocyclic compound-quinoline degradation and detoxification by microbial consortium: Integrated pathways, meta-transcriptomic analysis and toxicological evaluation. J. Hazard. Mater. 2024, 465, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.D.; Mao, G.N.; Liu, R.D.; Zhou, X.Z.; Bartlam, M.; Wang, Y.Y. Transcriptome Profiling of Stenotrophomonas sp. Strain WZN-1 Reveals Mechanisms of 2,2′,4,4′-Tetrabromodiphenyl Ether (BDE-47) Biotransformation. Environ. Sci. Technol. 2022, 56, 11288–11299. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhu, M.H.; Yuan, Y.B.; Rong, X.F.; Dang, Z.; Yin, H. Integrated toxicology, metabolomics, and transcriptomics analyses reveal the biodegradation and adaptation mechanisms to BDE-47 in Pseudomonas plecoglossicida. Chem. Eng. J. 2023, 454, 12. [Google Scholar] [CrossRef]
- Bezalel, L.; Hadar, Y.; Fu, P.P.; Freeman, J.P.; Cerniglia, C.E. Initial oxidation products in the metabolism of pyrene, anthracene, fluorene, and dibenzothiophene by the white rot fungus Pleurotus ostreatus. Appl. Environ. Microbiol. 1996, 62, 2554–2559. [Google Scholar] [CrossRef]
- Kamei, I.; Sonoki, S.; Haraguchi, K.; Kondo, R. Fungal bioconversion of toxic polychlorinated biphenyls by white-rot fungus, Phlebia brevispora. Appl. Microbiol. Biotechnol. 2006, 73, 932–940. [Google Scholar] [CrossRef]
- Sun, J.T.; Pan, L.L.; Zhu, L.Z. Formation of hydroxylated and methoxylated polychlorinated biphenyls by Bacillus subtilis: New insights into microbial metabolism. Sci. Total Environ. 2018, 613, 54–61. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | NCBI Accession No. (IMG ID) | Genome Size (Mb) | GC Content (%) |
|---|---|---|---|---|
| Kocuria rosea | 13 | CP127857.1 | 4.2 | 72.0 |
| Kocuria varians | 80 | CP059343.1 | 2.8 | 70.5 |
| Kocuria rhizophila | NBC_00781 | CP108927.1 | 2.8 | 71.0 |
| Kocuria flava | HO-9041 | CP013254.1 | 3.6 | 74.0 |
| Kocuria palustris | MU14/1 | CP012507.1 | 2.9 | 70.5 |
| Kocuria turfanensis | HO-9042 | CP012507.1 | 4.2 | 73.0 |
| Kocuria oceani | FXJ8.057 | JARAMH000000000.1 | 4.2 | 72.5 |
| Kocuria polaris | CMS 76or | JSUH00000000.1 | 3.8 | 73.0 |
| Kocuria sediminis | JCM 17929 | WOGU00000000.1 | 3.9 | 73.0 |
| Kocuria marina | AM104-93 | JAQDQP000000000.1 | 3.1 | 69.0 |
| Kocuria aegyptia | JCM 14735 | BAAAOA000000000.1 | 3.9 | 72.5 |
| Kocuria salina | CV6 | SSSF00000000.1 | 4.2 | 72.0 |
| Kocuria dechangensis | CGMCC 1.12187 | BMEQ00000000.1 | 4.5 | 72.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Nan, J. Comparative Genomic and Transcriptomic Analysis Provides New Insights into the Aflatoxin B1 Biodegradability by Kocuria rosea from Deep Sea. Microorganisms 2025, 13, 875. https://doi.org/10.3390/microorganisms13040875
Wang J, Nan J. Comparative Genomic and Transcriptomic Analysis Provides New Insights into the Aflatoxin B1 Biodegradability by Kocuria rosea from Deep Sea. Microorganisms. 2025; 13(4):875. https://doi.org/10.3390/microorganisms13040875
Chicago/Turabian StyleWang, Jingying, and Jun Nan. 2025. "Comparative Genomic and Transcriptomic Analysis Provides New Insights into the Aflatoxin B1 Biodegradability by Kocuria rosea from Deep Sea" Microorganisms 13, no. 4: 875. https://doi.org/10.3390/microorganisms13040875
APA StyleWang, J., & Nan, J. (2025). Comparative Genomic and Transcriptomic Analysis Provides New Insights into the Aflatoxin B1 Biodegradability by Kocuria rosea from Deep Sea. Microorganisms, 13(4), 875. https://doi.org/10.3390/microorganisms13040875