
Characterization and Genome Analysis of Fusarium oxysporum Provides Insights into the Pathogenic Mechanisms of the Pokkah Boeng Disease in China


 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Strain Culture and Morphological Observation
2.2. Pathogenicity Assay
2.3. Ultrastructure Analysis Using Scanning Electron Microscopy (SEM)
2.4. Reponses to Chemical Compounds In Vitro
2.5. Genome Sequencing, Assembly, and Evaluation
2.6. Gene Prediction
2.7. Gene Annotation
2.8. Phylogenetic Tree Construction
2.9. Comparative Genome Analysis
2.10. Transcriptome Analysis
3. Results
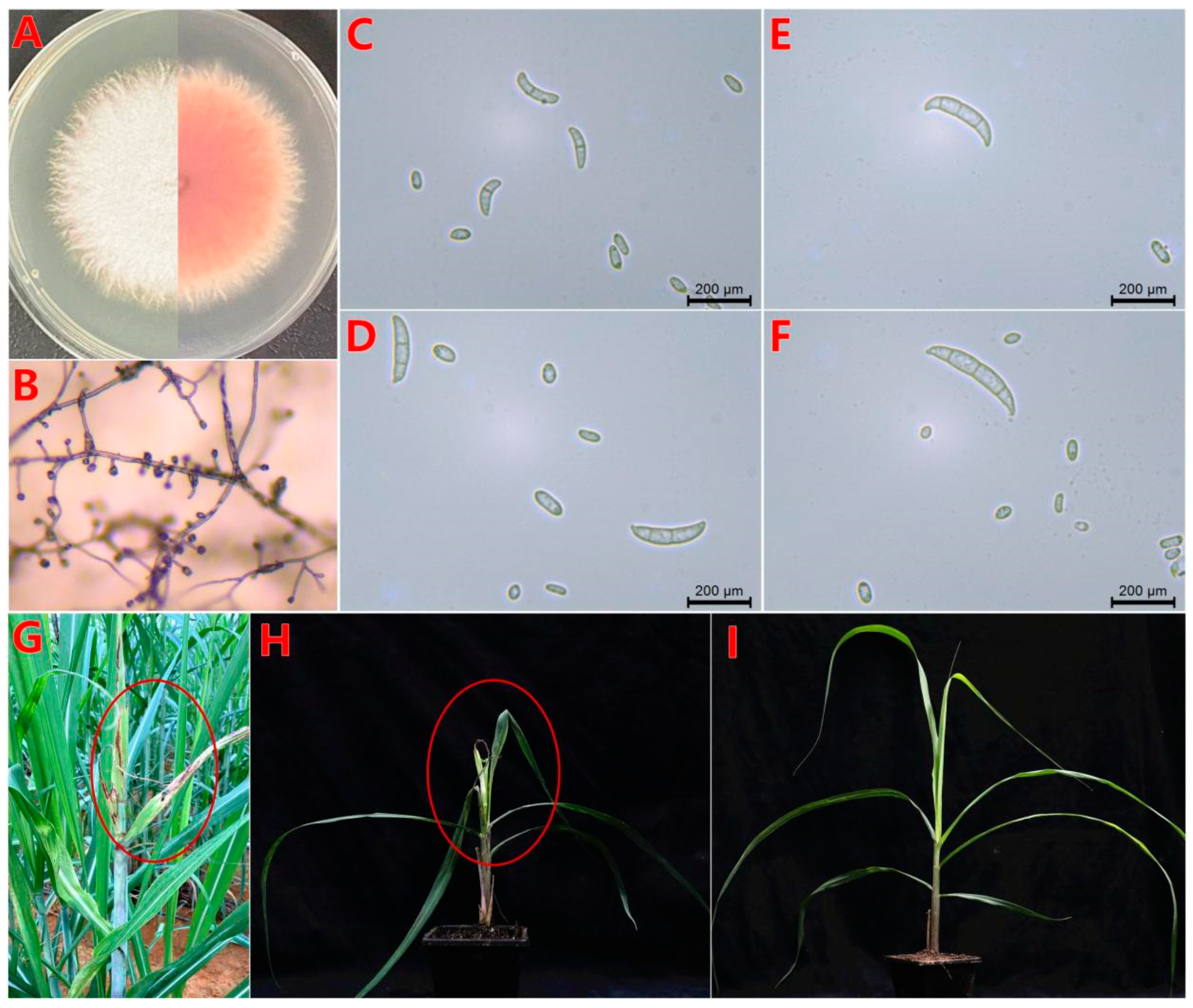
3.1. Colony Morphology and Determination of Pathogenicity
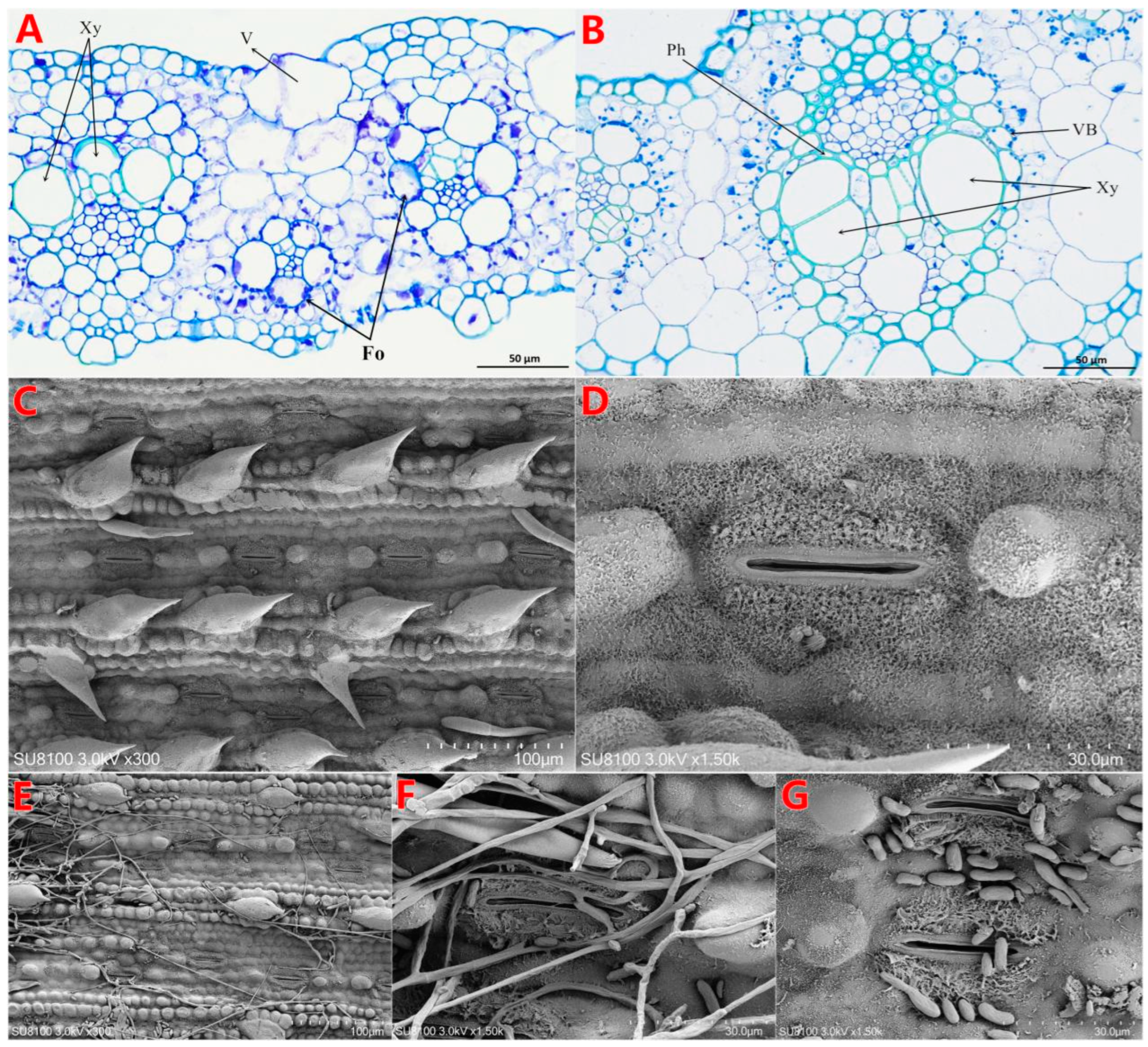
3.2. Effect of F. oxysporum BS2-6 on the Ultrastructure of Sugarcane Leaves
3.3. Physiological Response of F. oxysporum BS2-6 In Vitro
3.4. Sequencing, Assembly, and Evaluation
3.5. Genome Annotation
3.6. Phylogenetic Evolution
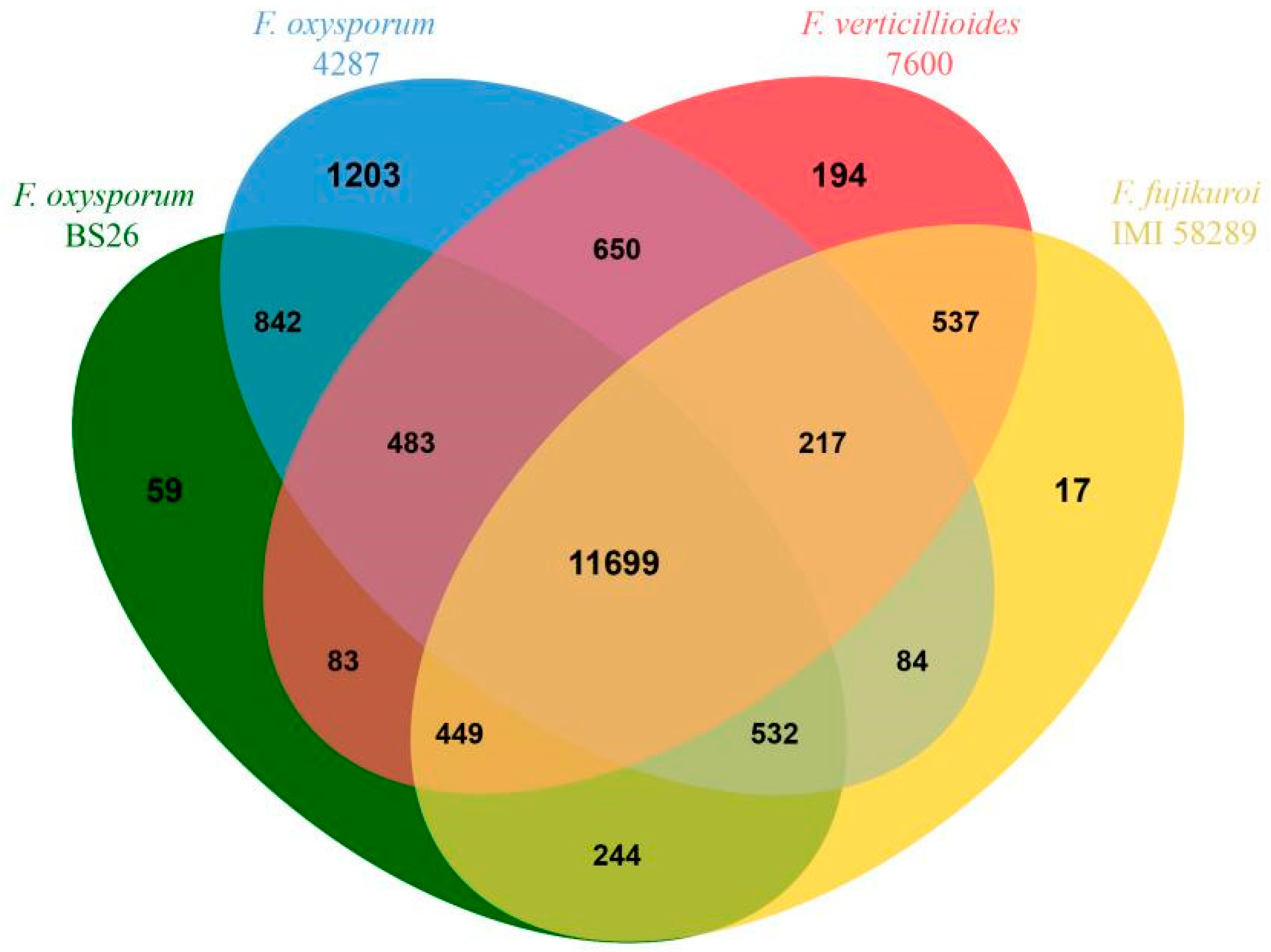
3.7. Comparative Genome Analysis
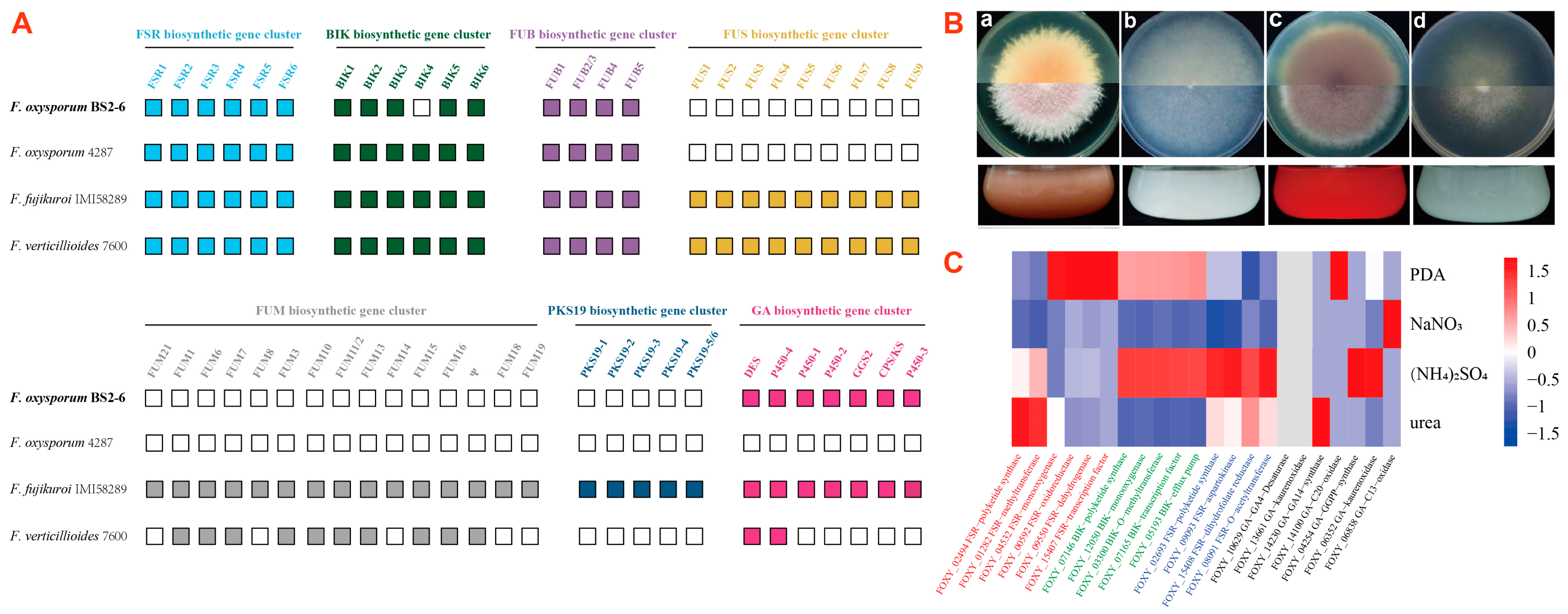
3.8. Identification of Secondary Metabolite Gene Families
3.9. Broad Spectrum Secondary Metabolite Production
3.10. Virulence Assay on Sugarcane
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, Z.Y.; Xu, S.Q.; Que, Y.X.; Wang, J.H.; Comstock, J.; Wei, J.J.; McCord, P.H.; Chen, B.S.; Chen, R.K.; Zhang, M.Q. Species-specific detection and identification of Fusarium species complex, the causal agent of sugarcane pokkah boeng in China. PLoS ONE 2014, 9, e104195. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.X.; Sehrish, A.; Yao, W.; Xu, Y.Z.; Xu, J.L.; Powell, C.A.; Chen, B.S.; Zhang, M.Q. Genetic diversity and pathogenicity of Fusarium fujikuroi species complex (FFSC) causing sugarcane pokkah boeng disease in China. Plant Dis. 2023, 107, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.X.; Sun, H.J.; Li, Y.F.; Duan, Z.Z.; McCord, P.H.; Cui, Y.P.; Zhang, M.Q. First Report of Fusarium oxysporum Isolate gx3 Causing Sugarcane Pokkah Boeng in Guangxi of China. Plant Dis. 2016, 100, 1785. [Google Scholar] [CrossRef]
- Armstrong, G.M.; Armstrong, J.K. Formae speciales and races of Fusarium oxysporum causing wilt diseases. In Fusarium Diseases Biology and Taxonomy; Pennsylvania State University Press: University Park, TX, USA, 1981; pp. 391–399. [Google Scholar]
- Burgess, L.W. General ecology of the fusaria. In Fusarium: Diseases, Biology and Taxonomy; Pennsylvania State University Press: University Park, TX, USA, 1981; pp. 225–235. [Google Scholar]
- Braun, C.J.; Jilka, J.M.; Hemenway, C.L.; Turner, N.E. Interactions between plants, pathogens and insects: Possibilities for engineering resistance. Curr. Opin. Biotechnol. 1991, 2, 193–198. [Google Scholar] [CrossRef]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Cellular, genomic and metabolic complexity. Biol. Rev. 2020, 95, 1198–1232. [Google Scholar] [CrossRef]
- Tedersoo, L.; Sánchez-Ramírez, S.; Kõljalg, U.; Bahram, M.; Döring, M.; Schigel, D.S.; May, T.W.; Ryberg, M.; Abarenkov, K. High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Divers. 2018, 90, 135–159. [Google Scholar] [CrossRef]
- Ma, L.J.; van der Does, C.H.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Pietro, A.D.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef]
- Yang, Y.R.; Li, Z.L.; Jin, M.H. How do chemical fertilizer reduction policies work? -Empirical evidence from rural China. Front. Environ. Sci. 2022, 10, 955278. [Google Scholar] [CrossRef]
- Lin, Z.Y.; Wang, J.H.; Bao, Y.X.; Guo, Q.; Powell, C.A.; Xu, S.Q.; Chen, B.S.; Zhang, M.Q. Deciphering the transcriptomic response of Fusarium verticillioides in relation to nitrogen availability and the development of sugarcane pokkah boeng disease. Sci. Rep. 2016, 6, 29692. [Google Scholar] [CrossRef]
- Afolayan, A.J.; Otunola, G.A. Ultrastructure and Elemental Analysis of Hypoxis Hemerocallidea: A Multipurpose Medicinal Plant. Afr. J. Tradit. Complement. Altern. Med. 2014, 11, 39–43. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Zhang, S.; Liu, X.Z.; Wen, H.A.; Wang, M. A simple method of genomic DNA extraction suitable for analysis of bulk fungal strains. Lett. Appl. Microbiol. 2010, 51, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 2019, 17, 155–158. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.; Zeng, Q.D.; Wortman, J.R.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, 265–268. [Google Scholar] [CrossRef]
- Han, Y.; Wessler, S. MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010, 38, e199. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, 351–358. [Google Scholar] [CrossRef]
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21, 152–158. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Seberg, O.; Petersen, G. A unified classification system for eukaryotic transposable elements should reflect their phylogeny. Nat. Rev. Genet. 2009, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Majoros, W.; Pertea, M.; Salzberg, S. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef]
- Haas, B.J.; Delcher, A.L.; Mount, S.; Wortman, J.R.; Smith, R.K.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.; Town, C.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Nawrocki, E.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.; Eddy, S.R.; Floden, E.W.; Gardner, P.P.; Jones, T.A.; Tate, J.; et al. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015, 43, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.E.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.; Natale, D.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated NR database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Saier, M.H.; Tran, V.C.; Barabote, R.D. TCDB: The Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006, 34, D181–D186. [Google Scholar] [CrossRef]
- Winnenburg, R.; Baldwin, T.; Urban, M.; Rawlings, C.J.; Köhler, J.; Hammond-Kosack, K. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L.; Bioinformatics, S. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Gardiner, D.M.; Dodds, P.N.; Tini, F.; Covarelli, L.; Singh, K.B.; Manners, J.; Taylor, J.M. EffectorP: Predicting Fungal Effector Proteins from Secretomes Using Machine Learning. New Phytol. 2016, 210, 743–761. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.H.; Rosli, H.G.; Pombo, M.; Zhang, P.F.; Banf, M.; Dai, X.B.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Team, A.C. Adobe Illustrator CS6 Classroom in a Book; Adobe Systems Incorporated: San Jose, CA, USA, 2012. [Google Scholar]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.A.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2012, 31, 46–54. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Wei, Y.B.; Jiang, C.X.; Pan, K.Y.; Wang, M.Y.; Deng, Z.H.; Zhang, M.Q. Occurrence and field evaluation of sugarcane genotypes for resistance to pokkah boeng disease in China. Plant Pathol. 2023, 72, 1088–1103. [Google Scholar] [CrossRef]
- Wiemann, P.; Sieber, C.M.K.; von Bargenm, K.W.; Studt, L.; Niehaus, E.M.; Espino, J.J.; Huß, K.; Michielse, C.B.; Albermann, S.; Wagner, D.; et al. Deciphering the Cryptic Genome: Genome-wide Analyses of the Rice Pathogen Fusarium fujikuroi Reveal Complex Regulation of Secondary Metabolism and Novel Metabolites. PLoS Pathog. 2013, 9, e1003475. [Google Scholar] [CrossRef] [PubMed]
- Booth, C. The Genus Fusarium; Commonwealth Mycological Institute: Kew, UK, 1971. [Google Scholar]
- Nelson, P.E.; Toussoun, T.A.; Marasas, W.F.O. Fusarium Species: An Illustrated Manual for Identification; Pennsylvania State University Press: University Park, TX, USA, 1983. [Google Scholar]
- Capelli, C.; Polverari, A.; Stravato, V.M. Fusarium oxysporum f. sp. melongenae on the eggplant. Inf. Fitopatol. 1993, 43, 51–54. [Google Scholar]
- Kistler, H.C. Evolution in Host Specificity in Fusarium oxysporum; Book Chapter; APS Press: Eagan, MN, USA, 2001. [Google Scholar]
- Jones, D.R. Fungal diseases of root, corm and pseudostem. In Handbook of Diseases of Banana Abac and Enset; CABI: Wallingford, UK, 2000; pp. 207–254. [Google Scholar] [CrossRef]
- Uehara, T.; Arase, S.; Honda, Y.; Park, P.; Nozu, M. Primary Effects of Magnaporthe grisea Toxin(s) on Mitochondria of Rice Leaf Cells. Jpn. J. Phytopathol. 1997, 63, 29–35. [Google Scholar] [CrossRef]
- Wang, Z.P.; Song, Q.; Liang, S.; Htun, R.; Malviya, M.K.; Li, Y.J.; Liang, Q.; Zhang, G.M.; Zhang, M.Q.; Zhou, F.J. Metabolic and proteomic analysis of nitrogen metabolism mechanisms involved in the sugarcane-Fusarium verticillioides interaction. J. Plant Physiol. 2020, 251, 153207. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Summary | BS2-6 |
|---|---|
| Genome size (bp) | 51,731,010 |
| Contig number | 31 |
| Contig N50 (bp) | 2,803,775 |
| Contig N90 (bp) | 1,130,085 |
| Contig max (bp) | 5,230,794 |
| GC content | 47.37% |
| Accession No. | JALBXV000000000 |
| Complete BUSCOs (C) | 289 (99.66%) |
| Complete and single-copy BUSCOs (S) | 288 (99.31%) |
| Complete and duplicated BUSCOs (D) | 1 (0.34%) |
| Fragmented BUSCOs (F) | 1 (0.34%) |
| Missing BUSCOs (M) | 0 |
| Total BUSCO groups searched | 290 |
| Method | Software | BS2-6 | |
|---|---|---|---|
| Ab initio | Augustus | - | 12,255 |
| Glimmer HMM | - | 14,455 | |
| SNAP | - | 14,877 | |
| Homolog-based | GeMoMa | F. fujikuroi IMI58289 | 14,581 |
| F. fujikuroi KSU3368 | 14,907 | ||
| F. fujikuroi FGSC8932 | 14,544 | ||
| Unigene | PASA | - | 17,684 |
| Integration | EVM | - | 15,794 |
| Gene density (number of genes per Mb) | 305 | ||
| Average gene length (Kb) | 1.6 | ||
| Total exon length (Mb) | 22.91 | ||
| Total intron length (Mb) | 2.5 | ||
| Exons | 45,417 | ||
| Average exon length (bp) | 504.4 | ||
| Introns | 29,623 | ||
| Average intron length (bp) | 84.29 | ||
| Database | F. oxysporum BS2-6 |
|---|---|
| GO | 8898 (56.34%) |
| KOG | 7339 (46.47%) |
| KEGG | 3774 (23.90%) |
| Pfam | 11,329 (71.73%) |
| Swiss-Prot | 8929 (56.53%) |
| TrEMBL | 15,729 (99.59%) |
| Nr | 15,729 (99.59%) |
| CAZyme | 917 (5.81%) |
| PHI | 5088 (32.21%) |
| TCDB | 154 (0.98%) |
| SP | 1243 (7.87%) |
| EP | 250 (1.58%) |
| tRNA | 338 (2.14%) |
| rRNA | 96 (0.61%) |
| Other ncRNA | 42 (0.27%) |
| Pseudogene | 27 (0.17%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, W.; Zhang, C.; Akbar, S.; Wu, S.; Yue, Y.; Wang, G.; Zhou, Y.; Powell, C.A.; Yao, W.; Xu, J.; et al. Characterization and Genome Analysis of Fusarium oxysporum Provides Insights into the Pathogenic Mechanisms of the Pokkah Boeng Disease in China. Microorganisms 2025, 13, 573. https://doi.org/10.3390/microorganisms13030573
Lin W, Zhang C, Akbar S, Wu S, Yue Y, Wang G, Zhou Y, Powell CA, Yao W, Xu J, et al. Characterization and Genome Analysis of Fusarium oxysporum Provides Insights into the Pathogenic Mechanisms of the Pokkah Boeng Disease in China. Microorganisms. 2025; 13(3):573. https://doi.org/10.3390/microorganisms13030573
Chicago/Turabian StyleLin, Wenfeng, Chi Zhang, Sehrish Akbar, Suyan Wu, Yabing Yue, Gege Wang, Yu Zhou, Charles A. Powell, Wei Yao, Jianlong Xu, and et al. 2025. "Characterization and Genome Analysis of Fusarium oxysporum Provides Insights into the Pathogenic Mechanisms of the Pokkah Boeng Disease in China" Microorganisms 13, no. 3: 573. https://doi.org/10.3390/microorganisms13030573
APA StyleLin, W., Zhang, C., Akbar, S., Wu, S., Yue, Y., Wang, G., Zhou, Y., Powell, C. A., Yao, W., Xu, J., Chen, B., Zhang, M., & Bao, Y. (2025). Characterization and Genome Analysis of Fusarium oxysporum Provides Insights into the Pathogenic Mechanisms of the Pokkah Boeng Disease in China. Microorganisms, 13(3), 573. https://doi.org/10.3390/microorganisms13030573