
Descriptive Comparative Transcriptomic Analysis of Genotype IV SHEV ORF3-Expressing HepG2 Cells


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Preparation of Recombinant Adenovirus AD-ORF3 and AD-GFP
2.3. RNA Sample Preparation and Transcriptome Sequencing
2.4. Bio-Informatics Analysis
2.5. qRT-PCR Validation
2.6. Comparative Analysis of Multiple Omics
3. Results
3.1. Transcriptome Sequencing Results
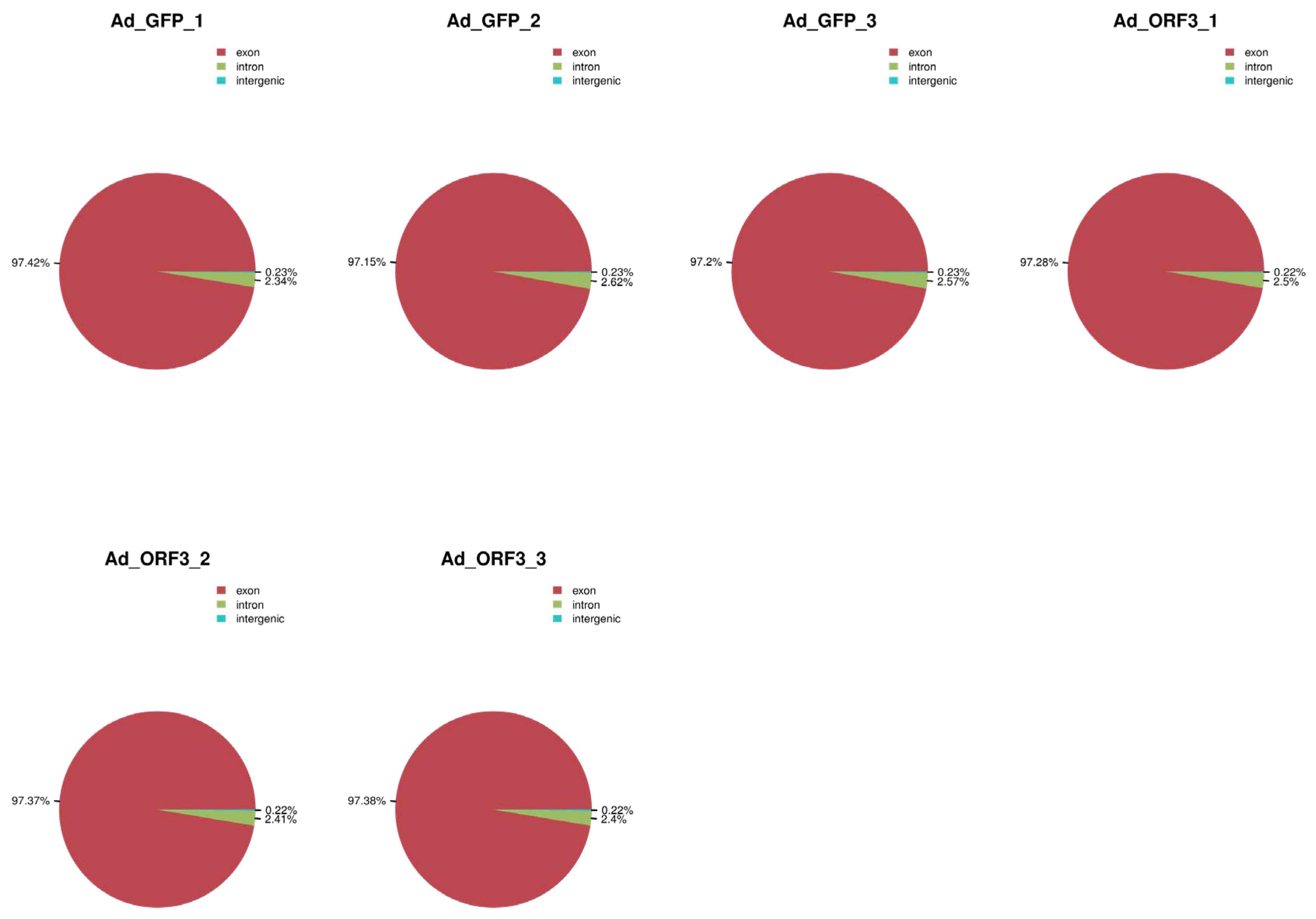
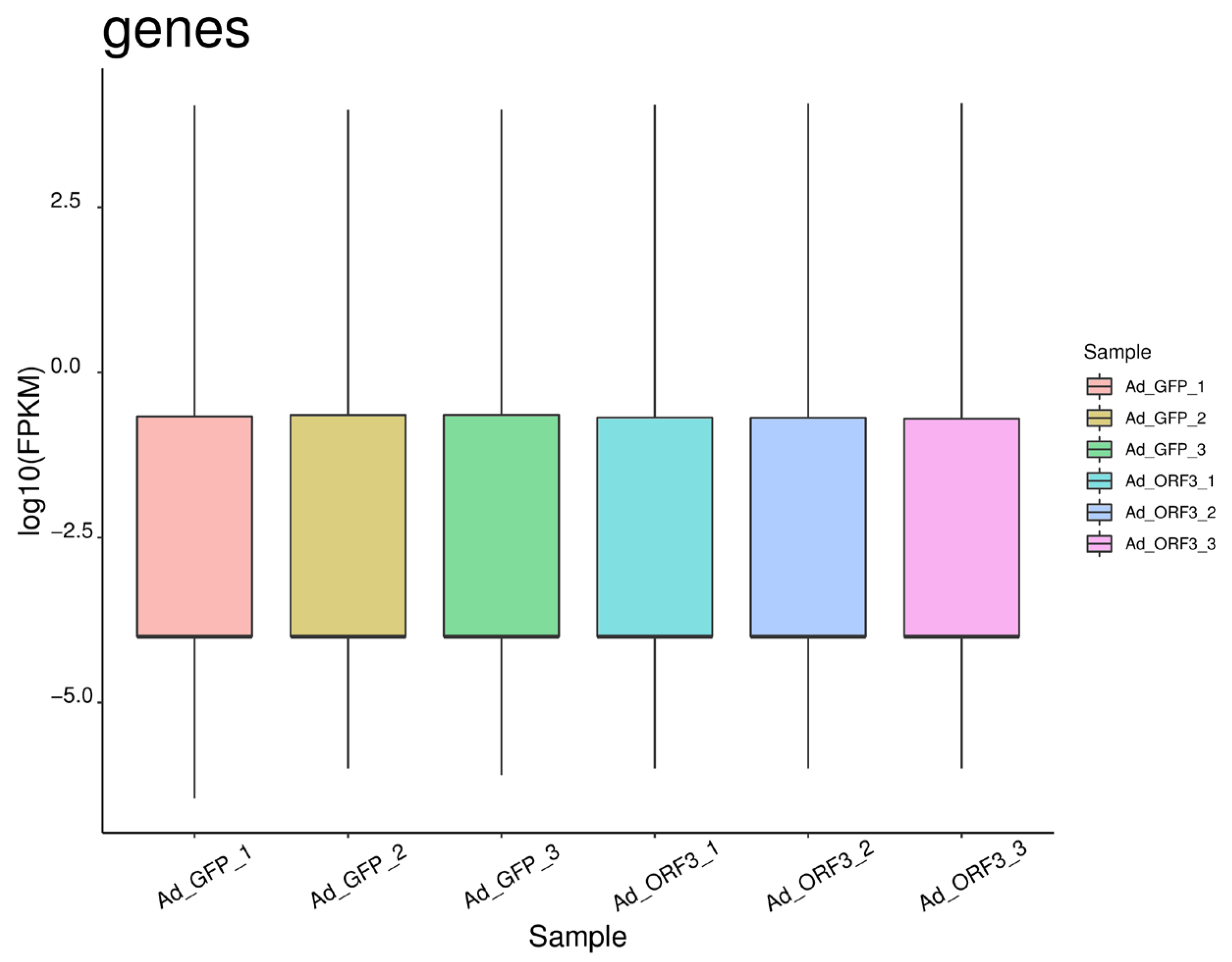
3.1.1. Overview of Transcriptome
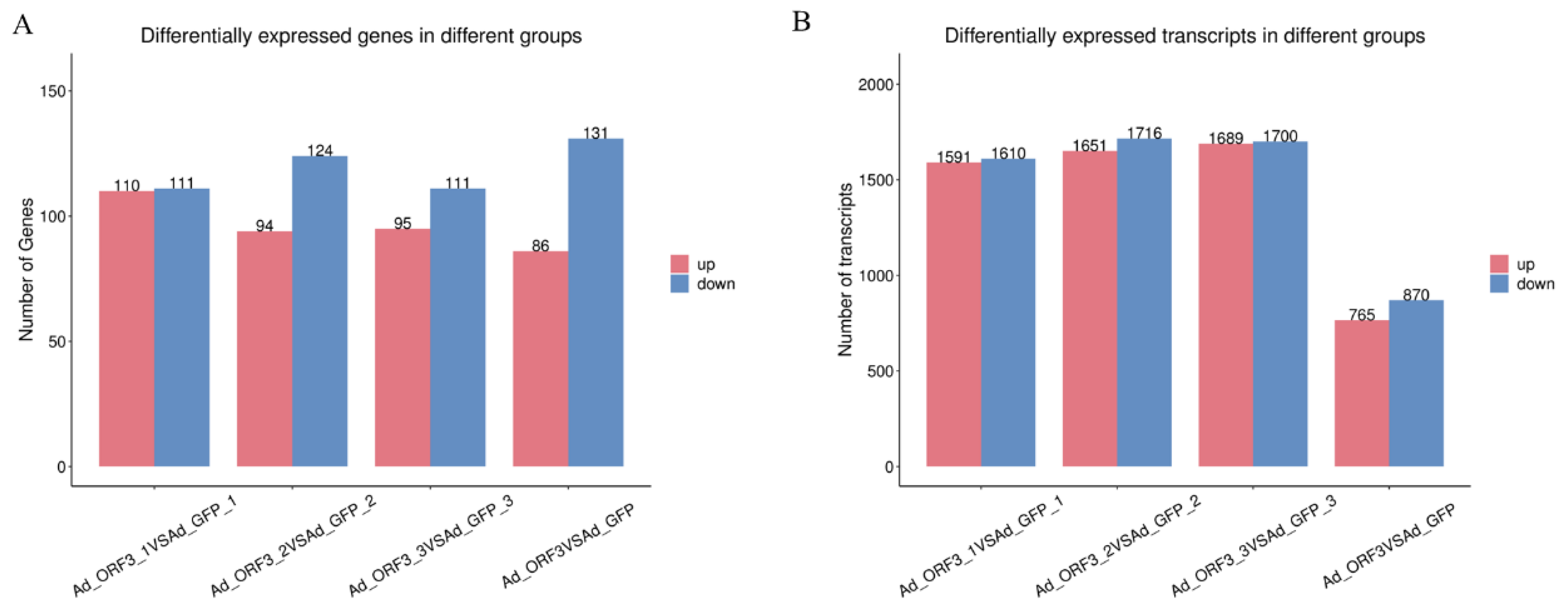
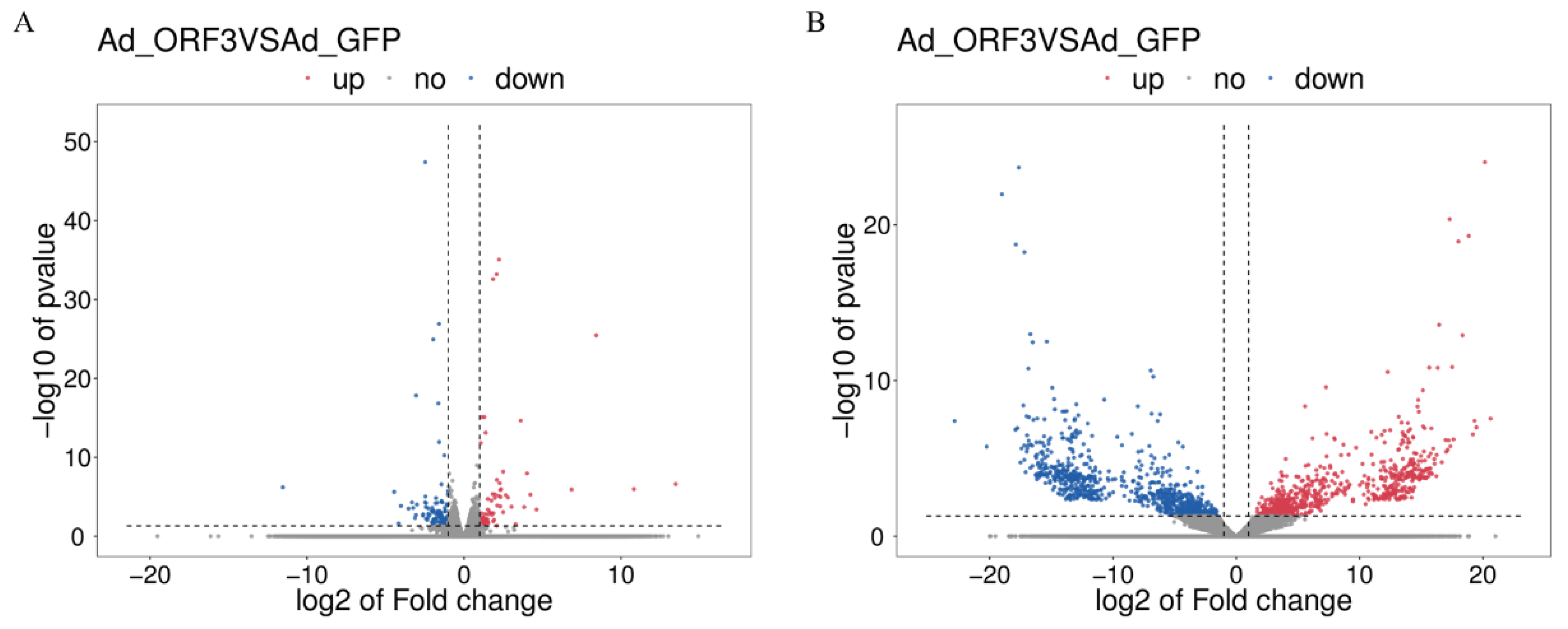
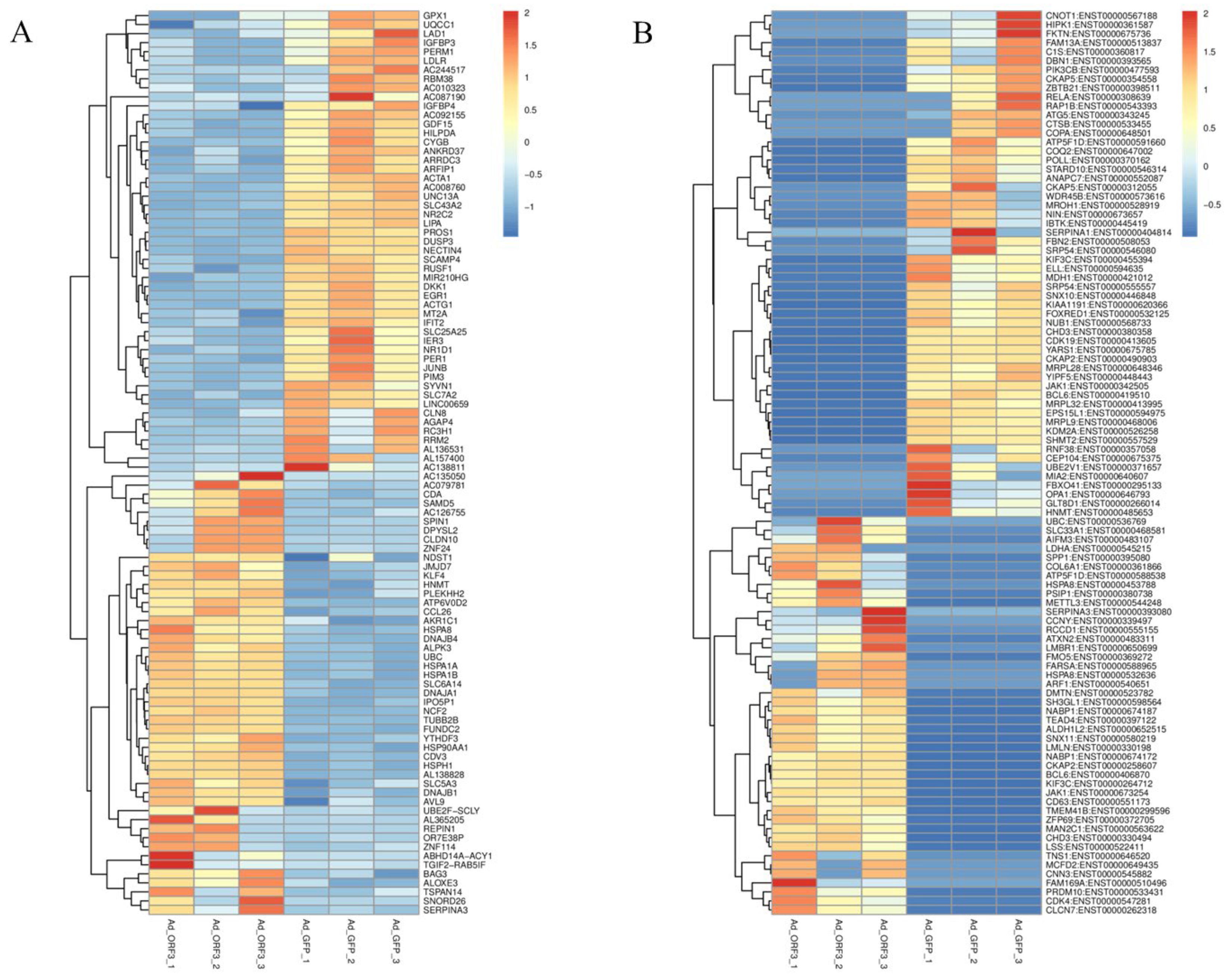
3.1.2. Differential Genes and Transcripts
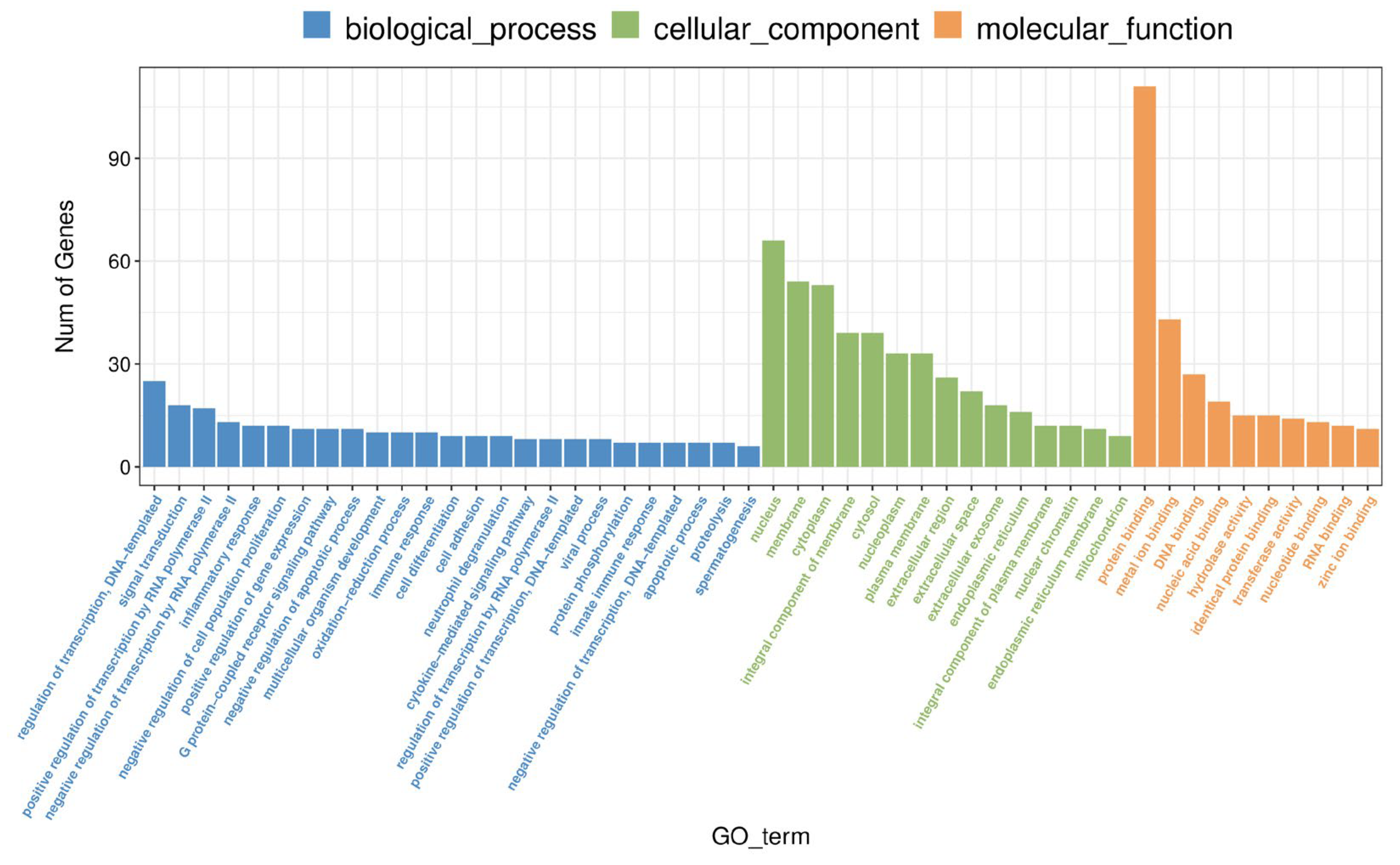
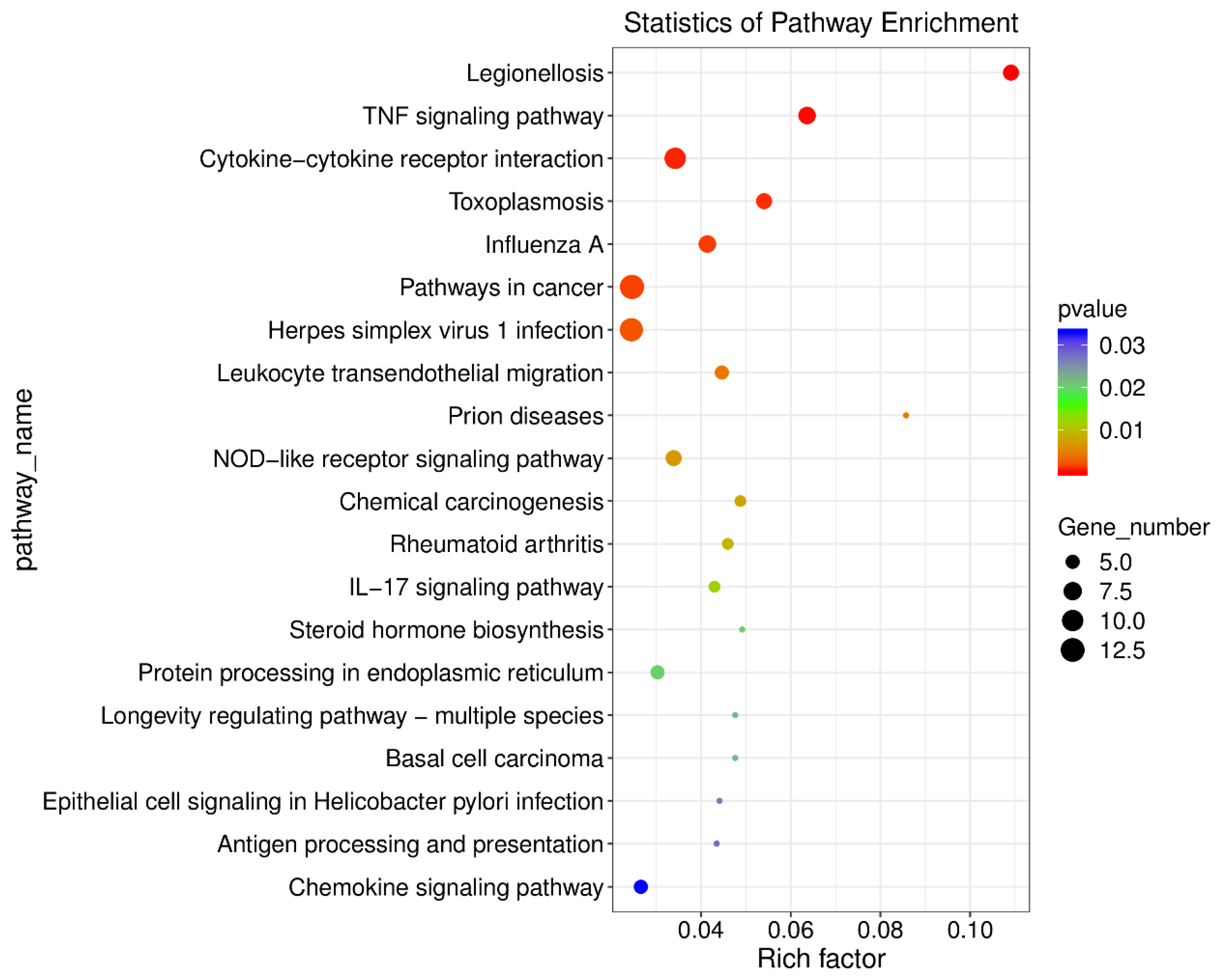
3.2. Further Screening of ORF3 Involvement in the Pathway
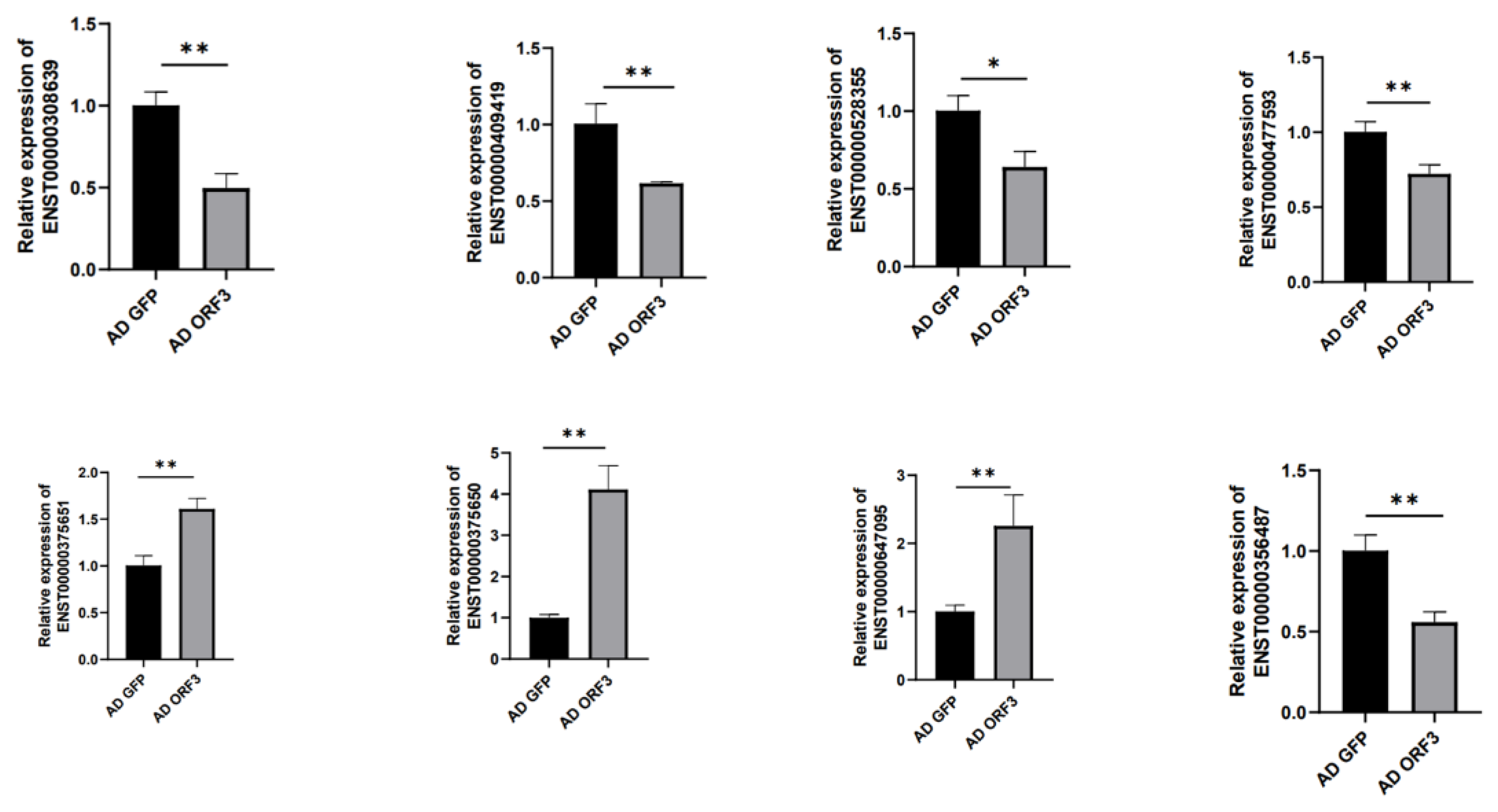
3.3. qRT-PCR
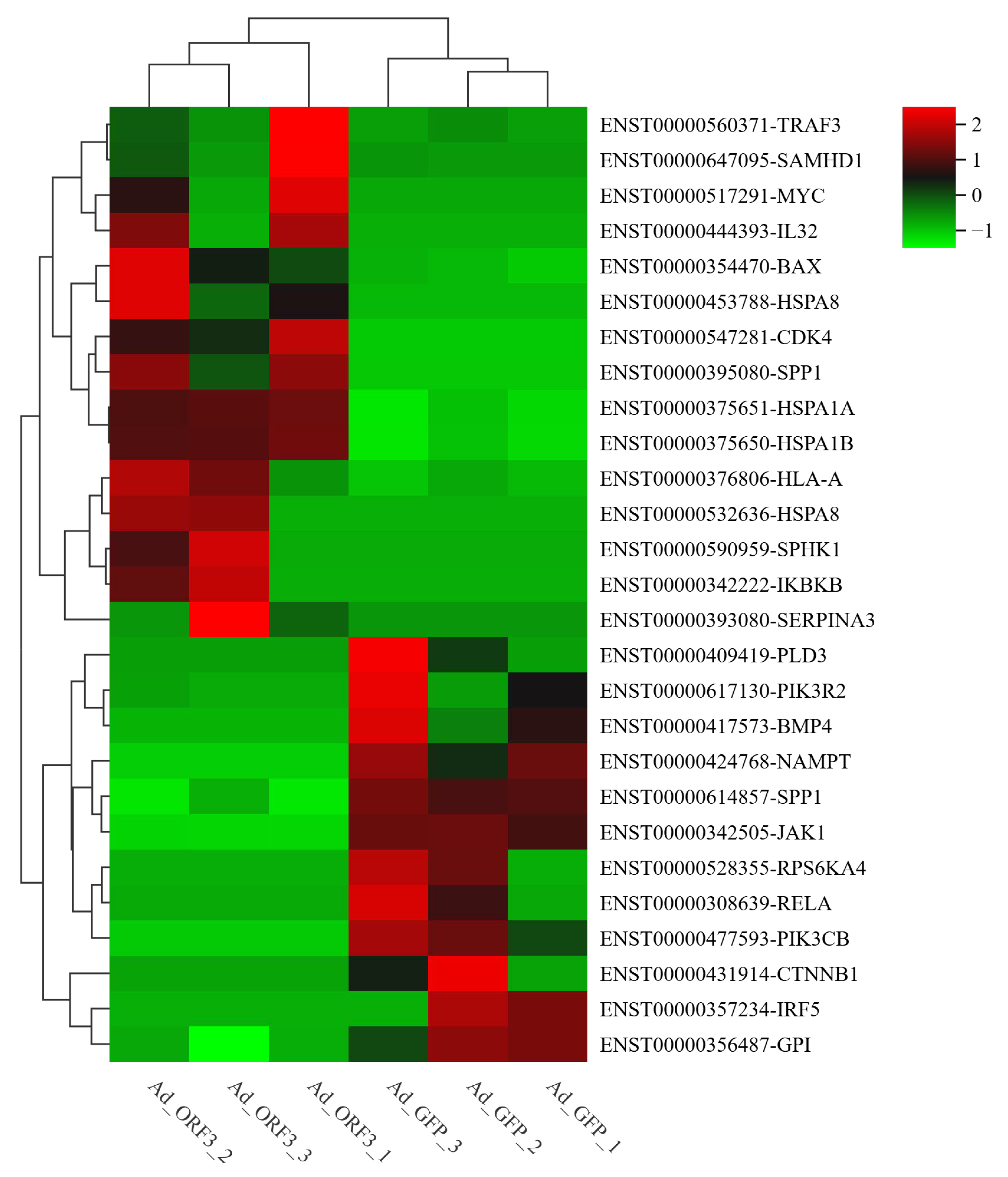
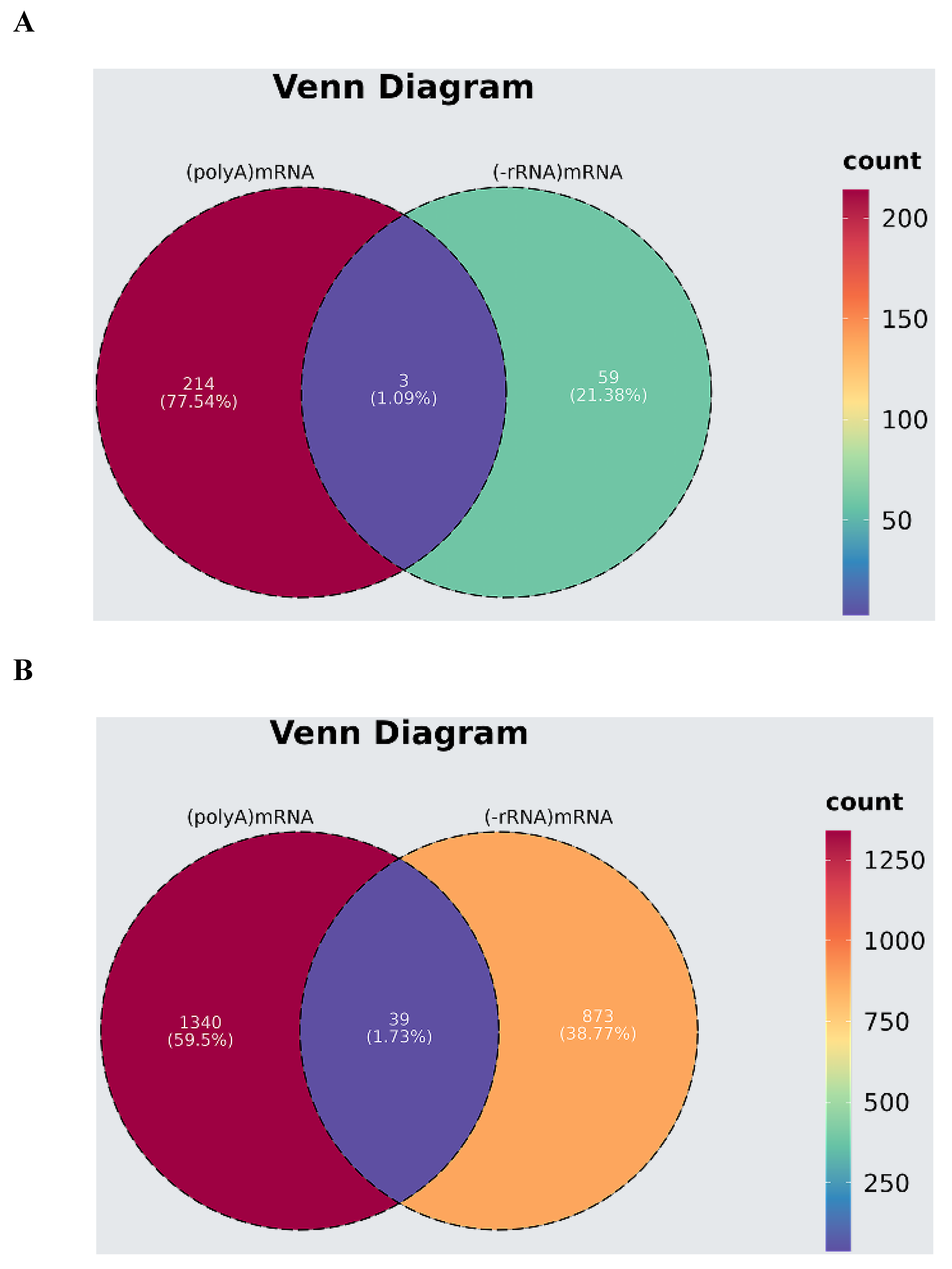
3.4. Comparative Analysis of Transcriptome and mRNA Expression Profiles of lncRNA Sequencing
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahmed, R.; Nasheri, N. Animal reservoirs for hepatitis E virus within the Paslahepevirus genus. Vet. Microbiol. 2023, 278, 109618. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J.; Purcell, R.H.; Halbur, P.G.; Lehman, J.R.; Webb, D.M.; Tsareva, T.S.; Haynes, J.S.; Thacker, B.J.; Emerson, S.U. A novel virus in swine is closely related to the human hepatitis E virus. Proc. Natl. Acad. Sci. USA 1997, 94, 9860–9865. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; He, Q.; Yan, L.; Li, M.; Liang, Z.; Shu, J.; Zhang, F.; Wang, L.; Wang, L. Infectivity and pathogenicity of different hepatitis E virus genotypes/subtypes in rabbit model. Emerg. Microbes Infect. 2020, 9, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Jiang, M.; Jin, M.; Qiu, Z.G.; Shen, Z.Q.; Cui, W.H.; Wang, D.N.; Gong, L.F.; Li, B.; Wang, X.W.; et al. Seroprevalence and evolutionary dynamics of genotype 4 hepatitis E virus in Shandong Province, China. World J. Gastroenterol. 2014, 20, 7955–7963. [Google Scholar] [CrossRef]
- Xu, L.D.; Zhang, F.; Xu, P.; Huang, Y.W. Cross-species transmission and animal infection model of hepatitis E virus. Microbes Infect. 2024, 27, 105338. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, X.; Cao, M.; Xu, H.; Liu, H.; Zhao, Q.; Zhou, E.-M.; Chen, Y.; Liu, B. Hepatitis E virus causes apoptosis of ovarian cells in hens and resulting in a decrease in egg production. Poult. Sci. 2024, 103, 103501. [Google Scholar] [CrossRef]
- Ralfs, P.; Holland, B.; Salinas, E.; Bremer, B.; Wang, M.; Zhu, J.; Ambardekar, C.; Blasczyk, H.; Walker, C.M.; Feng, Z.; et al. Soluble ORF2 protein enhances HEV replication and induces long-lasting antibody response and protective immunity in vivo. Hepatology 2023, 78, 1867–1881. [Google Scholar] [CrossRef]
- Wang, B.; Meng, X.-J. Structural and molecular biology of hepatitis E virus. Comput. Struct. Biotechnol. J. 2021, 19, 1907–1916. [Google Scholar] [CrossRef]
- Baha, S.; Zhang, M.; Behloul, N.; Liu, Z.; Wei, W.; Meng, J. Efficient production and characterization of immunogenic HEV-PCV2 chimeric virus-like particles. Vet. Microbiol. 2022, 268, 109410. [Google Scholar] [CrossRef]
- Wei, B.; Li, H.; Cheng, M.; Yang, Y.; Liu, B.; Tian, Y.; Sun, Y.; Liu, T.; She, R.; Tian, J. NLRP3 Inflammasome Activation Mediates Hepatitis E Virus-Induced Neuroinflammation. J. Viral Hepat. 2024, 31, 729–738. [Google Scholar] [CrossRef]
- Kobayashi, T.; Takahashi, M.; Ohta, S.; Hoshino, Y.; Yamada, K.; Jirintai, S.; Primadharsini, P.P.; Nagashima, S.; Murata, K.; Okamoto, H. Production and Characterization of Self-Assembled Virus-like Particles Comprising Capsid Proteins from Genotypes 3 and 4 Hepatitis E Virus (HEV) and Rabbit HEV Expressed in Escherichia coli. Viruses 2024, 16, 1400. [Google Scholar] [CrossRef] [PubMed]
- Bahoussi, A.N.; Guo, Y.; Wang, P.; Dahdouh, A.; Wu, C.; Xing, L. Genomic characteristics and recombination patterns of swine hepatitis E virus in China. Transbound. Emerg. Dis. 2022, 69, E3273–E3281. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Li, Y.; Xu, Z.; Zhao, J.; Li, F.; Deng, H.; Sun, X.; Zhu, L. Research on a rat model of genotype IV swine hepatitis E virus. Vet. Med. Sci. 2022, 8, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Cao, Q.M.; Subramaniam, S.; Yugo, D.M.; Heffron, C.L.; Rogers, A.J.; Kenney, S.P.; Tian, D.; Matzinger, S.R.; Overend, C.; et al. Pig model mimicking chronic hepatitis E virus infection in immunocompromised patients to assess immune correlates during chronicity. Proc. Natl. Acad. Sci. USA 2017, 114, 6914–6923. [Google Scholar] [CrossRef]
- Nemes, K.; Persson, S.; Simonsson, M. Hepatitis A Virus and Hepatitis E Virus as Food- and Waterborne Pathogens—Transmission Routes and Methods for Detection in Food. Viruses 2023, 15, 1725. [Google Scholar] [CrossRef]
- Ma, Z.; de Man, R.A.; Kamar, N.; Pan, Q. Chronic hepatitis E: Advancing research and patient care. J. Hepatol. 2022, 77, 1109–1123. [Google Scholar] [CrossRef]
- Geng, Y.; Shi, T.; Wang, Y. Transmission of Hepatitis E Virus. In Hepatitis E Virus; Wang, Y., Ed.; Springer Nature: Singapore, 2023; pp. 73–92. [Google Scholar] [CrossRef]
- Di Cola, G.; Fantilli, A.; Mamani, V.; Tamiozzo, P.; Wassaf, M.M.; Nates, S.V.; Ré, V.E.; Pisano, M.B. High circulation of hepatitis E virus (HEV) in pigs from the central region of Argentina without evidence of virus occurrence in pork meat and derived products. Res. Vet. Sci. 2023, 164, 105000. [Google Scholar] [CrossRef]
- Monini, M.; Ianiro, G.; De Sabato, L.; Bivona, M.; Ostanello, F.; Di Bartolo, I. Persistence of hepatitis E virus (HEV) subtypes 3c and 3e: Long-term cold storage and heat treatments. Food Microbiol. 2024, 121, 104529. [Google Scholar] [CrossRef]
- Lee, W.-J.; Kang, M.L.; Cha, S.-B.; Park, B.-K.; Choi, I.-S.; Yoo, H.S. Analysis of the helicase gene of Korean swine hepatitis E virus isolates and trends in viral infection. Arch. Virol. 2009, 154, 1361–1364. [Google Scholar] [CrossRef]
- Lee, G.; Han, D.; Song, J.Y.; Kim, J.H.; Yoon, S. Proteomic analysis of swine hepatitis E virus (sHEV)-infected livers reveals upregulation of apolipoprotein and downregulation of ferritin heavy chain. FEMS Immunol. Med. Microbiol. 2011, 61, 359–363. [Google Scholar] [CrossRef]
- Cheng, Y.; Du, L.; Shi, Q.; Jiao, H.; Zhang, X.; Hao, Y.; Rong, H.; Zhang, J.; Jia, X.; Guo, S.; et al. Identification of miR-221 and -222 as important regulators in genotype IV swine hepatitis E virus ORF3-expressing HEK 293 cells. Virus Genes 2013, 47, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Kannan, H.; Fan, S.; Patel, D.; Bossis, I.; Zhang, Y.-J. The hepatitis E virus open reading frame 3 product interacts with microtubules and interferes with their dynamics. J. Virol. 2009, 83, 6375–6382. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Shuai, X.; Luo, Y.; Zhou, Z.; Zhao, Y.; Li, B.; Gu, G.; Li, W.; Li, M.; Zeng, H.; et al. Deep Insight Into Long Non-coding RNA and mRNA Transcriptome Profiling in HepG2 Cells Expressing Genotype IV Swine Hepatitis E Virus ORF3. Front. Vet. Sci. 2021, 8, 625609. [Google Scholar] [CrossRef]
- Jiao, H.; Zhao, Y.; Zhou, Z.; Li, W.; Li, B.; Gu, G.; Luo, Y.; Shuai, X.; Fan, C.; Wu, L.; et al. Identifying Circular RNAs in HepG2 Expressing Genotype IV Swine Hepatitis E Virus ORF3 Via Whole Genome Sequencing. Cell Transplant. 2021, 30, 096368972110550. [Google Scholar] [CrossRef]
- Qian, Z.; Li, T.; Xia, Y.; Cong, C.; Chen, S.; Zhang, Y.; Gong, S.; Wang, W.; Liu, H.; Chen, D.; et al. Genotype 4 Hepatitis E virus replicates in the placenta, causes severe histopathological damage, and vertically transmits to fetuses. J. Infect. 2023, 87, 34–45. [Google Scholar] [CrossRef]
- Liu, T.; Cao, Y.; Weng, J.; Gao, S.; Jin, Z.; Zhang, Y.; Yang, Y.; Zhang, H.; Xia, C.; Yin, X.; et al. Hepatitis E virus infects human testicular tissue and Sertoli cells. Emerg. Microbes Infect. 2024, 13, 2332657. [Google Scholar] [CrossRef]
- Zhou, X.; Lu, H. Advances in Acute Severe Hepatitis of Unknown Etiology in Children. Zoonoses 2022, 2, 1–4. [Google Scholar] [CrossRef]
- Li, X.; Sun, X.; Pinpin, J.; Zhao, Q.; Sun, Y. Multifunctional ORF3 protein of hepatitis E virus. J. Med. Virol. 2024, 96, e29691. [Google Scholar] [CrossRef]
- Lei, Q.; Li, L.; Zhang, S.; Li, T.; Zhang, X.; Ding, X.; Qin, B. HEV ORF3 downregulates TLR7 to inhibit the generation of type I interferon via impairment of multiple signaling pathways. Sci. Rep. 2018, 8, 8585. [Google Scholar] [CrossRef]
- Xu, J.; Wu, F.; Tian, D.; Wang, J.; Zheng, Z.; Xia, N. Open Reading Frame 3 of Genotype 1 Hepatitis E Virus Inhibits Nuclear Factor-κappa B Signaling Induced by Tumor Necrosis Factor-α in Human A549 Lung Epithelial Cells. PLoS ONE 2014, 9, e100787. [Google Scholar] [CrossRef]
- Lei, Q.; Li, L.; Huang, W.; Qin, B.; Zhang, S. HEV ORF3 downregulatesCD14 and CD64 to impair macrophages phagocytosis through inhibiting JAK/STAT pathway. J. Med. Virol. 2019, 91, 1112–1119. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, L.; Wang, W.; Watashi, K.; Wang, Y.; Sprengers, D.; de Ruiter, P.E.; van der Laan, L.J.W.; Metselaar, H.J.; Kamar, N.; et al. Disparity of basal and therapeutically activated interferon signalling in constraining hepatitis E virus infection. J. Viral Hepat. 2016, 23, 294–304. [Google Scholar] [CrossRef]
- Salam, G.D.; Kumar, A.; Kar, P.; Aggarwal, S.; Husain, A.; Sharma, S. Serum tumor necrosis factor-alpha level in hepatitis E virus-related acute viral hepatitis and fulminant hepatic failure in pregnant women. Hepatol. Res. 2013, 43, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.-L.; Nie, L.-B.; Zou, Y.; Hou, J.-L.; Chen, X.-Q.; Bai, M.-J.; Gao, Y.-H.; Hu, G.-X.; Zhu, X.-Q. Serological evidence of hepatitis E virus and influenza A virus infection in farmed wild boars in China. Acta Trop. 2019, 192, 87–90. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Transcripts Name | Primer Sequence (5′-3′) |
|---|---|---|
| HSPA8 | ENST00000532636 | F: TCTTTCACCCAGTTCCCTGC |
| R: TGGACATGGTTGCCCCATAC | ||
| HSPA8 | ENST00000453788 | F: TTGGGTCTTGTAAGGGCAGC |
| R: GACATGGTTGCTGGGGTGTA | ||
| BAX | ENST00000354470 | F: CCCTTTTGCTTCAGGGGATGA |
| R: GAAGTCCAATGTCCAGCCCA | ||
| HSPA1A | ENST00000375651 | F: ATAAAAGCCCAGGGGCAAGC |
| R: AACACTGGATCCGCGAGAAG | ||
| HSPA1B | ENST00000375650 | F: GCAGGTGTGTAACCCCATCA |
| R: GAGTCCCAACAGTCCACCTC | ||
| SPP1 | ENST00000395080 | F: AGGCATCACCTGTGCCATAC |
| R: GGCCACAGCATCTGGGTATT | ||
| SERPINA3 | ENST00000393080 | F: TACATCCAGCTCCCTGAGAGT |
| R: TCAGGGGCCTTCAGGACTAA | ||
| SPHK1 | ENST00000590959 | F: CAGCACCGATAAGGAGCTGAA |
| R: AACCAAAGAGGCCACGTCC | ||
| PLD3 | ENST00000409419 | F: TGCCCACTTTTGTTCTGCCA |
| R: CCTTCCACGCCTCAATCTCA | ||
| RELA | ENST00000308639 | F: GCTCGTCTGTAGTGCACGC |
| R: TGATCTCCACATAGGGGCCA | ||
| IL32 | ENST00000444393 | F: ACCAGCTGAGTATTTGTGCCA |
| R: CGGCCAAAAGTTCAAGGAGC | ||
| GPI | ENST00000356487 | F: GTGTACCTTCTAGTCCCGCC |
| R: ATGCCCATGGTTGGTGTTGA | ||
| SPP1 | ENST00000614857 | F: AATTCTGGGAGGGCTTGGTT |
| R: TTTTCCTTGGTCGGCGTTTG | ||
| BMP4 | ENST00000417573 | F: AAGCCGAGGCGAGAGAGAC |
| R: CTCGGGATGGCACTACGGAA | ||
| NAMPT | ENST00000424768 | F: CCATGCCTCCAGTTTCGAGAT |
| R: TATCTGGGGGCAGTGATGGT | ||
| HLA-A | ENST00000376806 | F: CGGTGTATGGATTGGGGAGT |
| R: GCTTCTCTGGAAACCCGACA | ||
| SAMHD1 | ENST00000647095 | F: TGACATGTTCCACACTCGCA |
| R: ACTCTAGCCAAGTATCAAGGAAAA | ||
| JAK1 | ENST00000342505 | F: ATCCTGGAGCTGCAGACAGT |
| R: AGCTACTTCAGAGAAGCGCA | ||
| IRF5 | ENST00000357234 | F: CAGAGCTCAGCTTGGTCCC |
| R: GGATGGACTGGTTCATGGCA | ||
| IKBKB | ENST00000342222 | F: CGTGTCCTTCAGGGAGAGTG |
| R: TCCCCACGAATGATGTGCAA | ||
| PIK3R2 | ENST00000617130 | F: AGGCCATTGAAAGGACAGGG |
| R: GTGCCAGCAGGAAGCTCTTA | ||
| RPS6KA4 | ENST00000528355 | F: GAGGACGACGATGAGAGCTG |
| R: CTTGCCGTAGGCTTCTCCTC | ||
| PIK3CB | ENST00000477593 | F: TTGGAATAGTAGCAGGCGGC |
| R: CGCCCAGATGTCAAGGATGT | ||
| TRAF3 | ENST00000560371 | F: ACGGACCGCGAGAACTCC |
| R: CTTTAGCGGCGGGTTAGTCT | ||
| CDK4 | ENST00000547281 | F: GCTGGCGTGAGTGTACAAGG |
| R: CAGTCGCCTCAGTAAAGCCA | ||
| MYC | ENST00000517291 | F: CAAAAGAAAATGCAGCGGGC |
| R: CTAACGTTGAGGGGCATCGT | ||
| CTNNB1 | ENST00000431914 | F: CCCGGTGATTCAGGTCGAAA |
| R: GAGTAGCCATTGTCCACGCT |
| Sample | Raw Data | Valid Data | Valid Ratio (Reads) | Q20% | Q30% | GC Content % | ||
|---|---|---|---|---|---|---|---|---|
| Read | Base | Read | Base | |||||
| Ad_GFP_1 | 54,701,040 | 8.21 G | 53,701,618 | 8.06 G | 98.17 | 99.98 | 98.23 | 49 |
| Ad_GFP_2 | 49,681,178 | 7.45 G | 48,784,570 | 7.32 G | 98.20 | 99.98 | 98.48 | 49 |
| Ad_GFP_3 | 49,987,694 | 7.50 G | 49,061,438 | 7.36 G | 98.15 | 99.98 | 98.37 | 49 |
| Ad_ORF3_1 | 51,214,508 | 7.68 G | 50,288,760 | 7.54 G | 98.19 | 99.98 | 98.36 | 49 |
| Ad_ORF3_2 | 47,990,420 | 7.20 G | 47,125,072 | 7.07 G | 98.20 | 99.98 | 98.34 | 49 |
| Ad_ORF3_3 | 41,049,826 | 6.16 G | 40,340,186 | 6.05 G | 98.27 | 99.98 | 98.43 | 49 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiao, H.; Meng, C.; Jiao, F.; Zhou, G.; Wang, L.; Wu, S.; Fan, C.; Li, J.; Cao, L.; Zhao, Y.; et al. Descriptive Comparative Transcriptomic Analysis of Genotype IV SHEV ORF3-Expressing HepG2 Cells. Microorganisms 2025, 13, 412. https://doi.org/10.3390/microorganisms13020412
Jiao H, Meng C, Jiao F, Zhou G, Wang L, Wu S, Fan C, Li J, Cao L, Zhao Y, et al. Descriptive Comparative Transcriptomic Analysis of Genotype IV SHEV ORF3-Expressing HepG2 Cells. Microorganisms. 2025; 13(2):412. https://doi.org/10.3390/microorganisms13020412
Chicago/Turabian StyleJiao, Hanwei, Chi Meng, Fengyuan Jiao, Gengxu Zhou, Lingjie Wang, Shengping Wu, Cailiang Fan, Jixiang Li, Liting Cao, Yu Zhao, and et al. 2025. "Descriptive Comparative Transcriptomic Analysis of Genotype IV SHEV ORF3-Expressing HepG2 Cells" Microorganisms 13, no. 2: 412. https://doi.org/10.3390/microorganisms13020412
APA StyleJiao, H., Meng, C., Jiao, F., Zhou, G., Wang, L., Wu, S., Fan, C., Li, J., Cao, L., Zhao, Y., & Luo, Y. (2025). Descriptive Comparative Transcriptomic Analysis of Genotype IV SHEV ORF3-Expressing HepG2 Cells. Microorganisms, 13(2), 412. https://doi.org/10.3390/microorganisms13020412