Abstract
The virulence gene ToxA has been proposed to be horizontally transferred between three fungal wheat pathogens (Parastagonospora nodorum, Pyrenophora tritici-repentis, and Bipolaris sorokiniana) as part of a conserved ~14 kb ToxhAT transposon. Here, our analysis of 2137 fungal species-representative assemblies revealed that the ToxA gene is an isolate of Alternaria ventricosa and shows a remarkable 99.5% similarity to those found in B. sorokiniana and P. tritici-repentis. Analysis of the regions flanking ToxA within A. ventricosa revealed that it was embedded within a 14 kb genomic element nearly identical to the corresponding ToxhAT regions in B. sorokiniana, P. nodorum, and P. tritici-repentis. Comparative analysis further showed that ToxhAT in A. ventricosa resides within a larger mobile genetic element, which we identified as a member of the Starship transposon superfamily, named Frontier. Our analysis demonstrated that ToxhAT has been independently captured by three distinct Starships—Frontier, Sanctuary, and Horizon—which, despite having minimal sequence similarity outside of ToxhAT, facilitate its mobilization. These findings place Frontier, Sanctuary, and Horizon within a growing class of Starships implicated in the horizontal transfer of adaptive genes among fungal species. Moreover, we identified three distinct HGT events involving ToxA across these four fungal species, reinforcing the hypothesis of a single evolutionary origin for the ToxhAT transposon. These findings underscore the pivotal role of transposon-mediated HGT in the adaptive evolution of eukaryotic pathogens, offering new insights into how transposons facilitate genetic exchange and shape host–pathogen interactions in fungi.
1. Introduction
Horizontal gene transfer (HGT), the non-Mendelian exchange of genetic material between organisms, is a powerful force driving rapid adaptation, especially in microbial pathogens [1,2]. By enabling the acquisition of new traits, HGT can significantly enhance virulence and antibiotic resistance, expand host ranges, and improve competitive abilities in various environments [3,4,5]. Although HGT was once considered rare in eukaryotes, its impact on adaptation, particularly among microbes sharing hosts, is now increasingly recognized [1]. Fungi, with their relatively small genomes, critical roles in human and plant diseases, and extensive use in food and biotechnology, have emerged as ideal models for studying eukaryotic adaptation through HGT [6,7]. Recent studies have highlighted the widespread occurrence of HGT in fungi [8,9], even in complex multicellular forms like mushrooms [10]. Notably, the discovery of large transposable elements in fungal genomes, many of which harbor genes involved in HGT, underscores the crucial role these elements play in fungal evolution [11,12].
A prime example of HGT in fungi is the necrotrophic effector ToxA, initially identified in the wheat pathogen Pyrenophora tritici-repentis, where it induces severe lesions on susceptible wheat varieties [13,14]. Subsequently, ToxA and an associated 11 kb DNA region were also discovered in Parastagonospora nodorum, another wheat pathogen, which exhibits greater sequence diversity in ToxA compared to P. tritici-repentis [15]. This observation suggests that ToxA was horizontally transferred from P. nodorum to P. tritici-repentis, a hypothesis supported by the historical precedence of P. nodorum as a wheat pathogen [16]. Moreover, ToxA has been identified in Bipolaris sorokiniana, yet another wheat pathogen [17]. While all three species belong to the fungal order Pleosporales, they are relatively distant relatives, with their divergence occurring several million years ago [18]. This underscores the remarkable nature of HGT in shaping the evolutionary trajectory of these pathogens.
The horizontal transfer of ToxA between these species is thought to have been mediated by a Class II transposable element, now referred to as ToxhAT [19]. This 14 kb, single-copy Class II transposon is highly conserved across the three species, with ~92% nucleotide sequence similarity, indicating a recent HGT event [17]. While ToxhAT appears inactive in P. tritici-repentis and P. nodorum due to repeat-induced point (RIP) mutations that disrupt its terminal inverted repeats (TIRs), it remains largely intact in B. sorokiniana, likely due to minimal RIP activity [19,20].
Regions homologous to the DNA flanking ToxhAT, extending 61.2 kb upstream and 1.7 kb downstream, are shared between P. tritici-repentis and P. nodorum, suggesting that the horizontal transfer may have involved more than just ToxhAT [19]. Despite this, questions remain about the sequence of acquisition events and the origins of both ToxhAT and ToxA [21]. Notably, Gluck-Thaler et al. have identified a novel class of transposases termed “Starships,” characterized by a DUF3435-containing gene, which are capable of mobilizing large transposons [12]. Further work by Bucknell et al. revealed that the smaller ToxhAT transposon has been independently captured by two different Starships, namely Sanctuary in B. sorokiniana and Horizon in P. tritici-repentis and P. nodorum [22].
Although each of these species possesses additional pathogenicity mechanisms, the acquisition of ToxA is hypothesized to have played a pivotal role in their emergence as major pathogens of wheat. Understanding the mechanisms behind ToxA transfer and its retention within active transposons is therefore of paramount importance for the future management of these diseases. While ToxA has been well documented in Bipolaris sorokiniana, Parastagonospora nodorum, and Pyrenophora tritici-repentis, it has not yet been identified in the related Dothideomycetes, Alternaria ventricosa. A. ventricosa was first discovered as a new species on imported Ya Li pear fruit from China [23] and has also been isolated from wheat grains [24].
In this study, we report the discovery of ToxA in Alternaria ventricosa, marking its presence in this species for the first time. We compare this finding with other known species to further explore the mobility of ToxA, its associated transposon ToxhAT, and Starship elements. Our research provides valuable insights into the mechanisms underlying HGT and the evolutionary history of these transfer events, contributing to a better understanding of fungal adaptability and virulence.
2. Materials and Methods
2.1. Dataset
To explore the distribution of the ToxA gene across a broad range of fungal species, we analyzed a comprehensive dataset of 2137 whole-genome assemblies, which were sourced from the NCBI Reference Sequence Database as of July 2023. This dataset includes a diverse array of fungal genomes, with 1510 Ascomycetes, 476 Basidiomycetes, and 151 genomes from other fungal groups, providing a robust foundation for comparative analysis across multiple phylogenetic lineages. For detailed information on the specific species and genome assemblies used in this study, refer to Table S1.
2.2. Gene Prediction and Annotation
Gene prediction for the fungal genomes was carried out using GeneMark-ES [25] version 4.38, a tool specifically designed for eukaryotic genome annotation, with optimizations for fungal genomes to account for their unique intron–exon structures and complex intron organization. In addition to automated gene prediction, we conducted an extensive de novo annotation of repetitive elements across all three fungal species involved in this study. Manual annotation was employed for the regions surrounding the ToxA gene, ensuring a detailed and accurate characterization of the genomic context. This annotation facilitated a deeper understanding of the genomic environment in which the ToxA virulence gene is situated, particularly with respect to nearby transposable elements and other repeat families.
2.3. Sequence Comparisons
Amino acid sequence alignments of the ToxA genes from the selected species were performed using MAFFT [26] version 7.490, with default settings applied to ensure high accuracy and consistency in alignment quality. Following the alignment, a gene tree was constructed using IQ-TREE [27] version 2.0.3, utilizing its maximum likelihood (ML) framework to infer evolutionary relationships between ToxA sequences across the studied fungal species. Whole-genome average nucleotide identity (ANI) comparisons were carried out using pyANI version 0.2.9 software, enabling a quantitative assessment of genome-wide similarity between species containing ToxA. To evaluate the statistical significance of the horizontal gene transfer inference based on sequence similarity, we compared the observed genetic distances of the ToxA genes with the overall genome-wide ANI between the species. Assuming a Gaussian distribution of pairwise genetic distances, we calculated an exclusion range defined as μ ± 3σ, where μ is the mean and σ is the standard deviation of the genetic distances. Any similarity exceeding this threshold is considered statistically significant. The sequence similarity between ToxA genes observed in this study exceeded this threshold, providing strong statistical evidence supporting the HGT hypothesis. Scaffolds or chromosomes containing the ToxA gene were aligned across species using Progressive Mauve v2.3.1 [28], providing a comprehensive overview of synteny and structural variation in the genomic regions harboring ToxA. For the alignment of Starship transposons, visualizations were created using NGenomeSyn version 1.41 [29], which allowed for the identification and comparison of structural variants and insertion sites of mobile genetic elements.
2.4. Phylogenetic Tree of Species and HGT Detection
To assess genome completeness and ensure the inclusion of high-quality data, we used BUSCO v5.2.0 with default parameters, based on the presence or absence of 6641 predefined orthologs from the Pleosporales order (pleosporales_odb10 database). This quality control step was crucial for generating a robust phylogenomic dataset. A phylogenomic data matrix was constructed using the 6641 single-copy orthologs identified from representative species of Pleosporales. Each gene was aligned using MAFFT [26] version 7.490, employing default options to ensure high-quality alignments. Ambiguous regions within each alignment were trimmed using trimAl version 1.4 with the ‘gappyout’ option, further refining the data to focus on confidently aligned sequences.
The concatenated amino acid alignments of these BUSCO genes, each with greater than 50% taxon occupancy, were then used to build a comprehensive phylogenomic dataset. Phylogenetic analyses were performed using IQ-TREE [27] version 2.0.3, leveraging its maximum likelihood framework, which is well suited for large-scale phylogenomic datasets. The resulting phylogenetic tree, constructed in Newick format, was visualized using FigTree v1.4.0 and was drawn with a midpoint root to provide a balanced representation of evolutionary relationships (http://tree.bio.ed.ac.uk/software/figtree/ accessed on 1 May 2024). The majority of bootstrap values are 100%, indicating strong support for the tree’s topology.
To detect potential HGT events involving the ToxA gene, reconciliation analyses were conducted using RANGER-DTL (v2.0) [30], a specialized tool designed to infer gene duplication, loss, and transfer events. This method compares the ToxA gene tree with the species tree to identify discrepancies indicative of HGT.
3. Results
3.1. A Homolog of ToxA in the A. ventricosa Genome
We identified the ToxA gene on a 90.4 kb scaffold within the genome of Alternaria ventricosa BMP 2768, an isolate collected from pear in China in 2004 (NCBI BioSample: SAMN18144513). This discovery was made following an extensive analysis of 2137 species-representative fungal genomes. The genome of A. ventricosa was 34.7 Mbp in size, comparable to that of other species in the genus, such as A. alternata (33.0 Mbp), a common pathogen in a variety of natural food products. The A. ventricosa genome contains 12,891 predicted protein-coding genes, identified using GeneMark. To ensure the accuracy of our findings and rule out the possibility of contamination or errors, we reassembled the raw data from the FASTQ file using the SPAdes genome assembler. Upon examining the sequencing depth across the assembled contigs, we found that the scaffold containing the ToxA gene exhibited a depth consistent with that of other scaffolds, confirming the integrity and reliability of this genomic region.
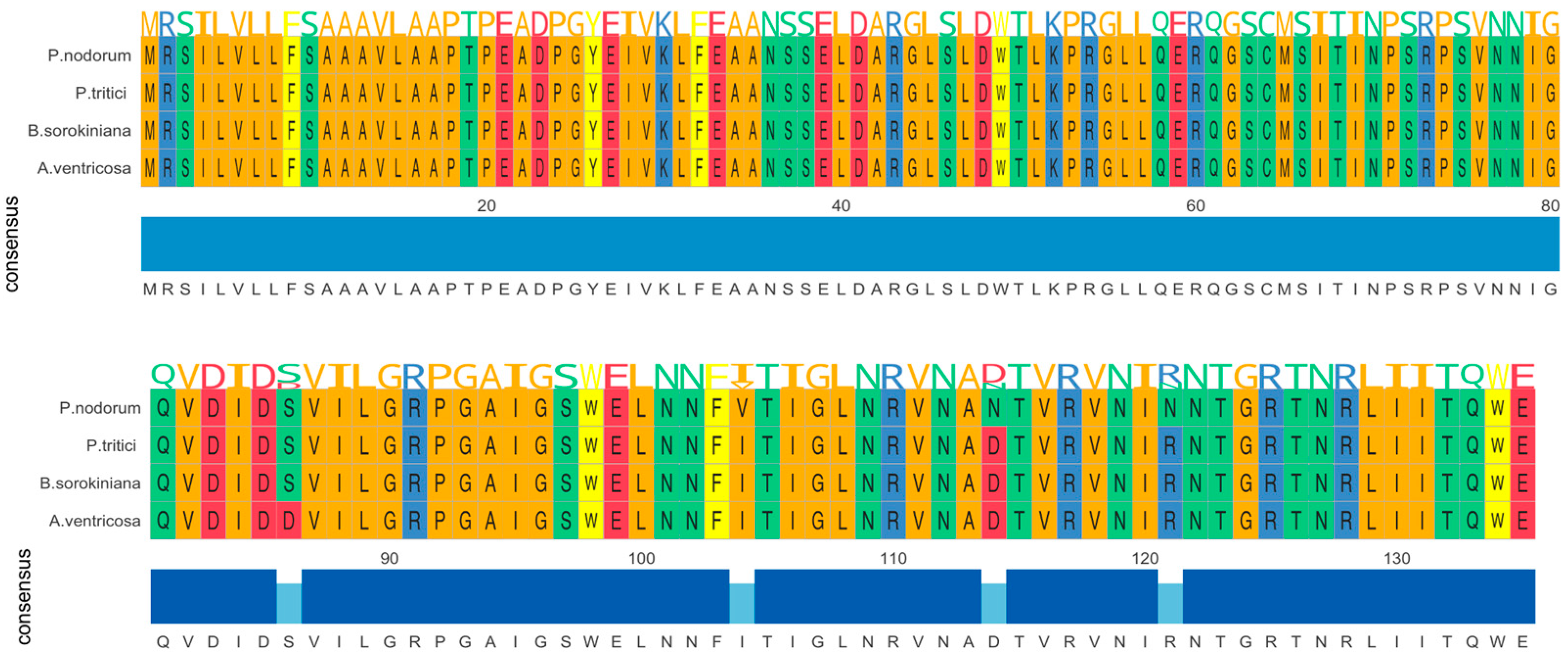
The ToxA gene in A. ventricosa shows remarkable sequence similarity to ToxA genes in Parastagonospora nodorum (98.0%), Bipolaris sorokiniana (99.5%), and Pyrenophora tritici-repentis (99.5%). Amino acid sequence alignment (Figure 1) reveals only four amino acid differences among these species. Whole-genome comparisons further indicated average nucleotide identity (ANI) values of 84.0%, 83.9%, and 84.0% with P. nodorum, B. sorokiniana, and P. tritici-repentis, respectively. Importantly, the identity between the ToxA genes and the ANI values showed a statistically significant difference (t-test, p < 0.01), emphasizing the distinct evolutionary processes of these genes compared to the overall genome sequences.
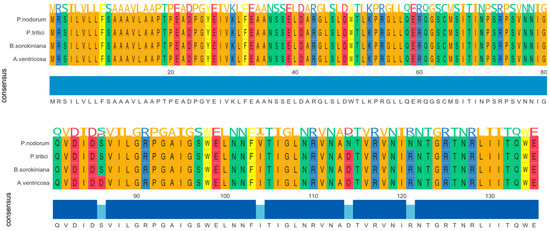
Figure 1.
Amono acid sequence alignment of ToxA genes from Alternaria ventricosa, Bipolaris sorokiniana, Parastagonospora nodorum, and Pyrenophora tritici-repentis. The alignment underscores the evolutionary relationships among these fungal pathogens and suggests that horizontal gene transfer of ToxA may have occurred between species of Pleosporales.
To further assess the significance of the observed similarity, we applied a statistical approach assuming a Gaussian distribution of pairwise genetic distances, with an exclusion range of μ ± 3σ. Based on this analysis, the similarity of 98% between the ToxA genes falls outside the μ + 3σ range, indicating that this high similarity is much higher than would be expected from the overall genome-wide similarity (p < 0.01). This further supports the hypothesis that the ToxA gene likely underwent horizontal gene transfer (HGT) between these species.
Importantly, no other known proteins showed significant similarity to ToxA (E-value < 0.00001), further underscoring the uniqueness of this gene. The high sequence similarity among ToxA genes from A. ventricosa, B. sorokiniana, P. nodorum, and P. tritici-repentis suggests a recent common ancestry, emphasizing the crucial role of horizontal gene transfer in the spread of this virulence factor across fungal species.
3.2. The 14 kbp ToxhAT Transposon in the A. ventricosa Genome
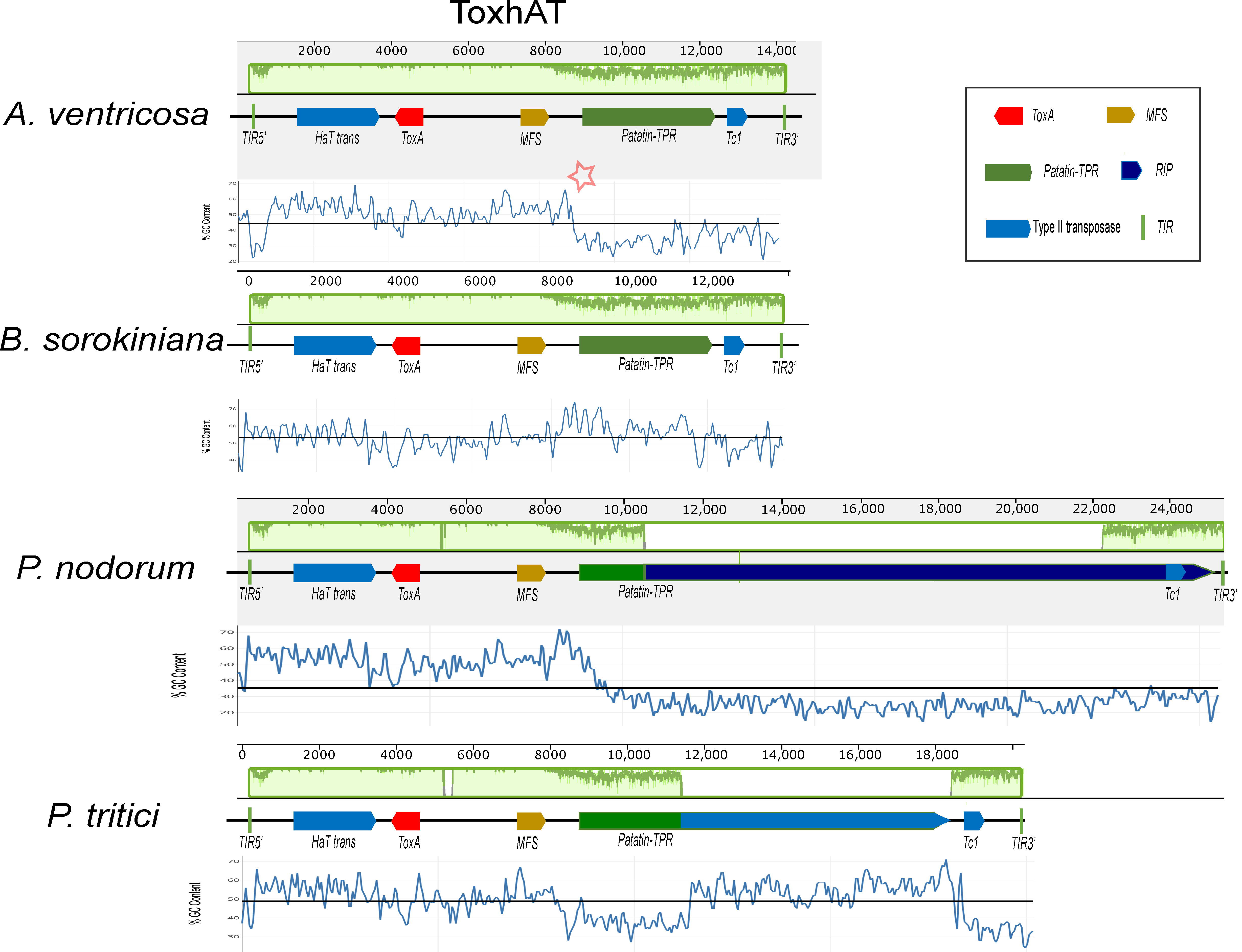
A conserved 14 kb genomic region, referred to as ToxhAT, was identified in A. ventricosa and compared across B. sorokiniana, P. nodorum, and P. tritici-repentis (Figure 2). Manual annotation of ToxhAT in each species revealed nucleotide identities of 93.5%, 94.8%, and 94.4% for A. ventricosa in comparison with B. sorokiniana, S. nodorum, and P. tritici-repentis, respectively. This 14 kb region aligns precisely with the region previously identified as horizontally transferred from P. nodorum to P. tritici-repentis, further supporting the hypothesis of HGT due to the striking nucleotide similarity across such a substantial DNA fragment.
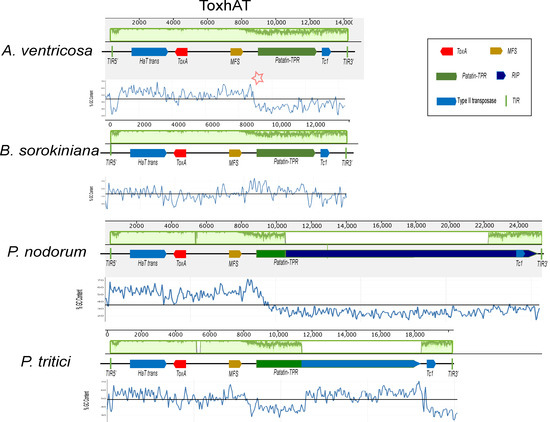
Figure 2.
Alignment of the ToxhAT region from Alternaria ventricosa, Pyrenophora tritici-repentis, Parastagonospora nodorum, and Bipolaris sorokiniana, generated using Mauve version 2.4.0. In the A. ventricosa genome, specific regions where AT richness increases are indicated by asterisks, highlighting the variation in sequence composition at the boundaries of the conserved 14 kb region. In the context of the Mauve alignment, this plot highlights the locally collinear blocks (LCBs) that are conserved across the genomes of the species being compared. Mauve is a multiple-genome alignment tool that identifies conserved genomic regions (LCBs) and visualizes them in the plot. The LCBs are typically aligned with high sequence similarity, and their conservation is highlighted in green. The green plot represents the regions of sequence conservation across the different species. The presence of Major Facilitator Superfamily (MFS) transporters, Patatin-like TPR domains (Patatin-TPR), and repeat-induced point (RIP) mutations is highlighted within the figure, as they are key to understanding the functional context of the ToxhAT region. The presence of terminal inverted repeats (TIRs) marks the ends of the transposon, providing further insight into its potential mobility across the genome. This alignment sheds light on the genomic architecture of ToxhAT and its potential evolutionary history.
Despite the high sequence identity within this 14 kb segment, there are key differences between the species, particularly toward the boundaries of the conserved region. As sequence similarity diminishes near the edges, there is a corresponding increase in AT richness. This pattern of increased AT content is predominantly due to the activity of repeat-induced point (RIP) mutations, particularly in P. nodorum and P. tritici-repentis. RIP, a fungal genome defense mechanism, targets repetitive elements and introduces mutations that degrade their functionality. These points of increased AT richness are highlighted for A. ventricosa in Figure 2 with asterisks, indicating potential RIP activity in this region as well.
3.3. Starship Elements Present in the A. ventricosa Genome
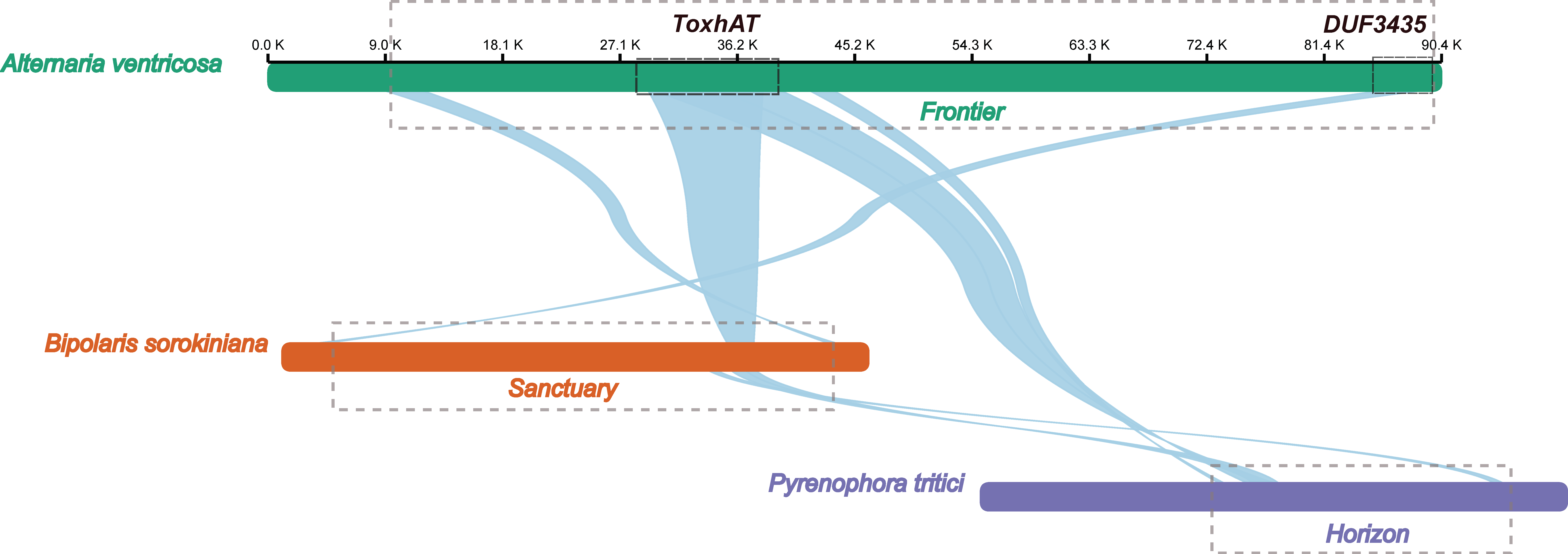
The 90.4 kb scaffold containing the ToxA gene from the A. ventricosa genome was aligned with the ToxA-containing regions from Bipolaris sorokiniana strain BRIP10943a (~200 kb), which includes the 196 kb Sanctuary element, and Pyrenophora tritici-repentis 1C-BFP (~200 kb), which includes the 143 kb Horizon element, using NGenomeSyn (Figure 3). The longest conserved collinear block spanned the 14 kb ToxhAT region. Notably, outside of the ToxhAT region, there was almost no significant sequence similarity between the Sanctuary and Horizon elements, underscoring the uniqueness of these regions.
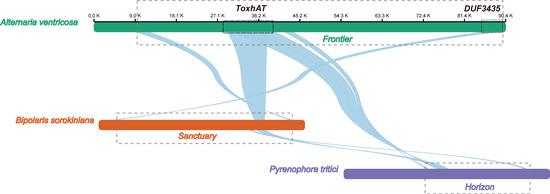
Figure 3.
Alignment of the ToxA-containing 90.4 kb scaffold from the A. ventricosa genome with the ToxA-containing regions from Bipolaris sorokiniana strain BRIP10943a (~200 kb), and Pyrenophora tritici-repentis 1C-BFP (~200 kb) using NGenomeSyn. This figure is crucial in highlighting the structural conservation and variation within ToxA loci and further supports the hypothesis of horizontal gene transfer events.
Interestingly, the first gene located at the 3′ end of the A. ventricosa scaffold was predicted to encode a DUF3435 domain protein. Alignment of the DUF3435 “captain” gene with B. sorokiniana revealed a high sequence identity of 98.1%. Further alignment of the 90.4 kb scaffold with Sanctuary showed that the first 3.8 kb, including the captain gene, was conserved with 98.5% nucleotide identity. Additionally, the 3.0 kb at the 5′ end, containing a predicted CHROMO domain protein implicated in histone binding, potentially enabling transposable elements to target specific genomic regions [31], exhibited 86.2% identity. Analysis of the genes within this transposon suggests that it is also a Starship element, sharing key structural features such as the DUF3435 tyrosine recombinase “captain” and ankyrin repeats (PF12796 and PF00023). Given these similarities, this newly identified Starship element, spanning 81.9 kb between the captain domain protein and the CHROMO domain protein, was named ‘Frontier’. However, beyond these conserved regions and homologous genes, Frontier shared limited sequence similarity with Sanctuary and Horizon. This conservation of terminal regions, along with the captain and CHROMO domain protein, may represent the minimal unit required for transposition.
A BLASTn search revealed that Frontier is not present in any species outside of A. ventricosa, highlighting its unique distribution. Comparative analysis across all three fungal species known to carry ToxhAT indicates that this transposon was independently captured by three distantly related Starships—Frontier in A. ventricosa, Sanctuary in B. sorokiniana, and Horizon in P. tritici-repentis. These findings underscore the complex evolutionary dynamics of Starship transposons and their role in horizontal gene transfer among fungi.
3.4. Interspecies HGT Events of ToxA
To explore the potential role of HGT in the discontinuous distribution of the ToxA gene across fungal species, we employed established methodologies, particularly focusing on reconciliation analysis using the RANGER-DTL program. This program is specifically designed to infer HGT, gene duplication, and loss events by reconciling discrepancies between gene trees and species trees.
For our analysis, we first constructed a species tree by examining 43 representative species within the order Pleosporales, providing a robust phylogenetic framework. We then generated a ToxA gene tree and reconciled it with the species tree using RANGER-DTL, which revealed three distinct interspecies HGT events (Figure 4).
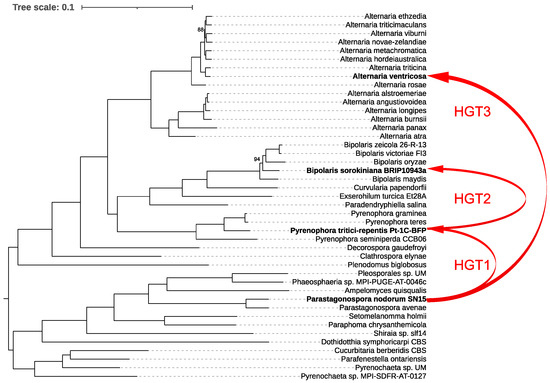
Figure 4.
Phylogenetic relationships and horizontal gene transfer (HGT) of ToxA among 43 representative species within the order Pleosporales. The phylogenetic tree was constructed using IQ-TREE based on a comprehensive alignment of 6641 conserved monocore genes. The schematic representation highlights the inferred horizontal transfers of the ToxA gene between fungal species, as indicated by the red lines mapped onto the phylogenetic tree. These red lines illustrate all detected HGT events of ToxA across the species, emphasizing the complex and widespread nature of these transfers within Pleosporales. Bootstrap values below 100% are shown; values of 100% were omitted for clarity.
The first event involved the transfer of ToxA from Parastagonospora nodorum to Pyrenophora tritici-repentis. Following this, P. tritici-repentis subsequently transferred ToxA to Bipolaris sorokiniana. The third HGT event identified was the transfer of ToxA from P. nodorum to Alternaria ventricosa. These findings underscore the complex evolutionary history of ToxA and highlight the significant role of HGT in shaping the pathogenic potential of these fungal species.
4. Discussion
ToxA has been demonstrated previously to play a dominant role in the wheat diseases caused by B. sorokiniana, P. nodorum, and P. tritici-repentis [15]. Our study extends this understanding by identifying ToxA in Alternaria ventricosa, a species not previously associated with wheat pathogenicity. This discovery underscores the broad role of ToxA in facilitating disease across a diverse range of fungal pathogens. Notably, the ToxA gene is part of a mobile “pathogenicity element” within these fungi, suggesting that this genetic region plays a key role in pathogen adaptability and virulence. The high mobility of ToxA, supported by its location within a transposon-rich region, raises the possibility of its rapid dissemination across fungal species, thereby enhancing their pathogenic potential on wheat and possibly other crops.
Alternaria is a ubiquitous fungal genus that includes saprobic, endophytic, and pathogenic species [32]. It is associated with a wide variety of substrates including seeds, plants, agricultural products, animals, soil, and the atmosphere. Species of Alternaria are known as serious plant pathogens, causing major losses in a wide range of crops [32]. Several taxa are also important postharvest pathogens, causative agents of phaeohyphomycosis in immuno-compromised patients, or airborne allergens. Alternaria ventricosa is increasingly recognized as a potential plant pathogen, and the presence of the ToxA gene further suggests its role in pathogenicity. This broad ecological distribution and the presence of ToxA in a new species of Alternaria highlight the complex evolutionary dynamics of fungal virulence and the potential for A. ventricosa to impact agriculture in ways not previously anticipated.
Despite the high sequence similarity across the conserved 14 kb region, notable differences exist between the four species. As sequence similarity diminishes towards the edges of this region, there is a corresponding increase in AT richness, primarily driven by the activity of repeat-induced point (RIP) mutations in P. nodorum and P. tritici-repentis. RIP, a well-characterized fungal genome defense mechanism, protects against the proliferation of repetitive elements [20]. During meiosis, the RIP machinery identifies invasive repetitive DNA and introduces CpG to CpA mutations, which often leads to the insertion of stop codons in coding regions, thereby disrupting transposon activity [33]. The increase in AT content at specific loci in Alternaria ventricosa is highlighted by asterisks in Figure 2, reflecting its distinct sequence composition relative to the other species. However, the exact role of ToxhAT as an active transposon in A. ventricosa remains speculative, warranting further investigation into its potential mobilization and the functional dynamics of this genomic region.
Previous studies have suggested multiple HGT events for ToxA and the surrounding 14 kb region, known as the ToxhAT transposon [15]. ToxhAT, a smaller transposon, has been independently captured by two distinct Starships, Sanctuary in B. sorokiniana and Horizon in P. tritici-repentis and P. nodorum [22]. In this study, we identified the nesting of ToxhAT within a larger putative 81.9 kb Starship transposon, which we named Frontier. Unlike Sanctuary and Horizon, Frontier is not present in other ToxA-containing species. While all three Starships—Frontier, Sanctuary, and Horizon—carry ToxhAT, they share minimal sequence similarity outside this region, indicating that ToxhAT was independently captured by each Starship. The mechanisms driving the ‘capture’ of cargo genes like ToxhAT into Starships remain poorly understood. Nevertheless, the presence of this key virulence factor within three distinct Starships adds to the growing body of evidence suggesting that Starships facilitate HGT between fungi, and that the cargo they transport provides a fitness advantage to the fungal host [12,34].
The origins of ToxhAT remain enigmatic. Since the initial discovery of ToxA in the genome of P. nodorum, the evolutionary history of this gene has been a subject of ongoing debate [15,35,36]. P. nodorum currently exhibits the highest known sequence diversity for ToxA, lending support to the hypothesis that ToxA has resided in its genome for the longest period, allowing ample time for the accumulation of mutations [15,36]. The subsequent identification of ToxA in B. sorokiniana and the characterization of conserved 74 bp terminal inverted repeats across all three species strongly suggest that ToxhAT had a single evolutionary origin in these fungi. Our study further elucidates this evolutionary pathway, identifying three distinct interspecies HGT events (Figure 4). The first event confirms that ToxA was horizontally transferred from P. nodorum to P. tritici-repentis, aligning with earlier findings. This initial transfer appears to have set the stage for subsequent HGT events, where P. tritici-repentis transferred ToxA to B. sorokiniana. Finally, our analysis uncovered a third HGT event, where ToxA was transferred from P. nodorum to A. ventricosa.
These findings not only reinforce the hypothesis of a single evolutionary origin for ToxhAT but also highlight the dynamic and ongoing role of HGT in the dissemination of this virulence gene across different fungal species. The presence of ToxA in a wide variety of fungal pathogens underscores the importance of transposon-mediated HGT in facilitating rapid genetic exchange, enabling fungi to adapt quickly to new host plants and environmental pressures. In particular, transposons may act as vehicles for the mobilization of virulence genes like ToxA, facilitating their spread across ecologically distinct fungal populations. This phenomenon exemplifies how transposon-driven HGT can significantly contribute to the adaptive evolution of fungi, particularly in the context of host–pathogen interactions, where rapid evolution is crucial for pathogens’ survival and success.
Looking ahead, several exciting avenues for future research arise from these findings. First, unraveling the specific mechanisms behind transposon-mediated gene capture and transfer will be crucial for understanding the ecological dynamics of ToxA and similar virulence genes. Investigating how these transposons are mobilized, the factors that influence their acquisition by different fungal species, and the ecological conditions that favor their spread will help us better predict the emergence and spread of new pathogenic strains. Furthermore, the study of transposon activity [34] and HGT in other agriculturally important fungal species will enhance our ability to track the movement of virulence factors in fungal populations, informing disease control strategies and potentially identifying targets for novel antifungal treatments.
In addition, the integration of genomic and ecological data could help identify key environmental factors that influence the rate of HGT in fungal populations. For example, how climate change, crop rotation practices, or the use of fungicides might alter fungal gene flow and the spread of virulence factors remains an area ripe for investigation [37]. Additionally, understanding the role of transposon activity in shaping fungal genome architecture could provide new insights into the long-term evolution of fungal pathogens and their adaptability to agricultural systems.
Ultimately, by expanding our understanding of how transposons facilitate the transfer of virulence genes, this research has the potential to revolutionize strategies for managing fungal diseases. Whether by developing more effective disease-resistant crops, improving diagnostic methods for detecting virulent strains, or creating more targeted fungicides, the implications for agricultural practices and crop protection are significant. By bridging the gap between basic fungal biology, transposon evolution, and practical disease management, these insights could lead to more sustainable agricultural practices and enhanced resilience to fungal pathogens in the face of changing environmental conditions.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/microorganisms13020376/s1. Table S1: The 2137 whole-genome assemblies analyzed in the study.
Author Contributions
Methodology, F.L.; formal analysis, F.L.; investigation, F.L.; writing—original draft preparation, F.L.; writing—review and editing, F.L.; visualization, F.L.; supervision, R.C. and R.-L.Z.; funding acquisition, R.C. and R.-L.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Project ID: 31961143010, 31970010, 32300012).
Data Availability Statement
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Huang, J. Horizontal Gene Transfer in Eukaryotes: The Weak-Link Model. Bioessays 2013, 35, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Palmer, J.D. Horizontal Gene Transfer in Eukaryotic Evolution. Nat. Rev. Genet. 2008, 9, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, X.; Xu, C.; Zhao, H.; Zhang, X.; Zeng, G.; Qian, Y.; Liu, R.; Guo, N.; Mi, W.; et al. Horizontal Gene Transfer Allowed the Emergence of Broad Host Range Entomopathogens. Proc. Natl. Acad. Sci. USA 2019, 116, 7982–7989. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Liu, F.; Liu, S.; Wang, J.; Zhu, B.; Shi, Y.; Gao, G.F. Identification of Antibiotic Resistance Genes and Associated Mobile Genetic Elements in Permafrost. Sci. China Life Sci. 2021, 64, 2210–2213. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhu, Y.; Yi, Y.; Lu, N.; Zhu, B.; Hu, Y. Comparative Genomic Analysis of Acinetobacter Baumannii Clinical Isolates Reveals Extensive Genomic Variation and Diverse Antibiotic Resistance Determinants. BMC Genom. 2014, 15, 1163. [Google Scholar] [CrossRef] [PubMed]
- Marcet-Houben, M.; Gabaldón, T. Acquisition of Prokaryotic Genes by Fungal Genomes. Trends Genet. 2010, 26, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-X.; Opulente, D.A.; Kominek, J.; Zhou, X.; Steenwyk, J.L.; Buh, K.V.; Haase, M.A.B.; Wisecaver, J.H.; Wang, M.; Doering, D.T.; et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 2018, 175, 1533–1545.e20. [Google Scholar] [CrossRef]
- Aleksander, M. The Interkingdom Horizontal Gene Transfer in 44 Early Diverging Fungi Boosted Their Metabolic, Adaptive, and Immune Capabilities. Evol. Lett. 2024, 8, 526–538. [Google Scholar]
- Liu, F.; Wang, S.-H.; Cheewangkoon, R.; Zhao, R.-L. Uneven Distribution of Prokaryote-Derived Horizontal Gene Transfer in Fungi: A Lifestyle-Dependent Phenomenon. mBio 2024, 16, e0285524. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ma, X.-B.; Han, B.; Wang, B.; Xu, J.-P.; Cao, B.; Ling, Z.-L.; He, M.-Q.; Zhu, X.-Y.; Zhao, R.-L. Pan-Genome Analysis Reveals Genomic Variations during Enoki Mushroom Domestication, with Emphasis on Genetic Signatures of Cap Color and Stipe Length. J. Adv. Res. 2024; in press. [Google Scholar] [CrossRef]
- Bucknell, A.H.; McDonald, M.C. That’s No Moon, It’s a Starship: Giant Transposons Driving Fungal Horizontal Gene Transfer. Mol. Microbiol. 2023, 120, 555–563. [Google Scholar] [CrossRef]
- Gluck-Thaler, E.; Ralston, T.; Konkel, Z.; Ocampos, C.G.; Ganeshan, V.D.; Dorrance, A.E.; Niblack, T.L.; Wood, C.W.; Slot, J.C.; Lopez-Nicora, H.D.; et al. Giant Starship Elements Mobilize Accessory Genes in Fungal Genomes. Mol. Biol. Evol. 2022, 39, msac109. [Google Scholar] [CrossRef] [PubMed]
- Tuori, R.P.; Wolpert, T.J.; Ciuffetti, L.M. Purification and Immunological Characterization of Toxic Components from Cultures of Pyrenophora Tritici-Repentis. Mol. Plant Microbe Interact. 1995, 8, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Ciuffetti, L.M.; Francl, L.J.; Ballance, G.M.; Bockus, W.W.; Lamari, L.; Meinhardt, S.W.; Rasmussen, J.B. Standardization of Toxin Nomenclature in the Pyrenophora Tritici-Repentis/Wheat Interaction. Can. J. Plant Pathol. 1998, 20, 421–424. [Google Scholar] [CrossRef]
- Friesen, T.L.; Stukenbrock, E.H.; Liu, Z.; Meinhardt, S.; Ling, H.; Faris, J.D.; Rasmussen, J.B.; Solomon, P.S.; McDonald, B.A.; Oliver, R.P. Emergence of a New Disease as a Result of Interspecific Virulence Gene Transfer. Nat. Genet. 2006, 38, 953–956. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.C.; Oliver, R.P.; Friesen, T.L.; Brunner, P.C.; McDonald, B.A. Global Diversity and Distribution of Three Necrotrophic Effectors in Phaeosphaeria Nodorum and Related Species. New Phytol. 2013, 199, 241–251. [Google Scholar] [CrossRef]
- McDonald, M.C.; Ahren, D.; Simpfendorfer, S.; Milgate, A.; Solomon, P.S. The Discovery of the Virulence Gene ToxA in the Wheat and Barley Pathogen Bipolaris Sorokiniana. Mol. Plant Pathol. 2017, 19, 432–439. [Google Scholar] [CrossRef]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse Lifestyles and Strategies of Plant Pathogenesis Encoded in the Genomes of Eighteen Dothideomycetes Fungi. PLoS Pathog. 2012, 8, e1003037. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.C.; Taranto, A.P.; Hill, E.; Schwessinger, B.; Liu, Z.; Simpfendorfer, S.; Milgate, A.; Solomon, P.S. Transposon-Mediated Horizontal Transfer of the Host-Specific Virulence Protein ToxA between Three Fungal Wheat Pathogens. mBio 2019, 10, e01515-19. [Google Scholar] [CrossRef]
- Galagan, J.E.; Selker, E.U. RIP: The Evolutionary Cost of Genome Defense. Trends Genet. 2004, 20, 417–423. [Google Scholar] [CrossRef]
- Ghaderi, F.; Sharifnabi, B.; Javan-Nikkhah, M.; Brunner, P.C.; McDonald, B.A. SnToxA, SnTox1, and SnTox3 Originated in Parastagonospora Nodorum in the Fertile Crescent. Plant Pathol. 2020, 69, 1482–1491. [Google Scholar] [CrossRef]
- Bucknell, A.; Wilson, H.M.; Gonçalves Do Santos, K.C.; Simpfendorfer, S.; Milgate, A.; Germain, H.; Solomon, P.S.; Bentham, A.; McDonald, M.C. Sanctuary: A Starship Transposon Facilitating the Movement of the Virulence Factor ToxA in Fungal Wheat Pathogens. bioRxiv 2024. [Google Scholar] [CrossRef]
- Roberts, R.G.; Roberts, R.G. Two New Species of Alternaria from Pear Fruit. Mycotaxon 2007, 100, 159–167. [Google Scholar]
- Fadhil, W.F.; Al-Saadoon, A.H.; Al-Moussawi, F.M. New Records of Mycobiota Associated with Stored Wheat and Its By- Products in Iraq. Biodiversitas 2022, 23, 3099–3107. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene Prediction in Novel Fungal Genomes Using an Ab Initio Algorithm with Unsupervised Training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Yang, J.; Jing, Y.; Xu, L.; Yu, K.; Fang, X. NGenomeSyn: An Easy-to-Use and Flexible Tool for Publication-Ready Visualization of Syntenic Relationships across Multiple Genomes. Bioinformatics 2023, 39, btad121. [Google Scholar] [CrossRef]
- Bansal, M.S.; Kellis, M.; Kordi, M.; Kundu, S. RANGER-DTL 2.0: Rigorous Reconstruction of Gene-Family Evolution by Duplication, Transfer and Loss. Bioinformatics 2018, 34, 3214–3216. [Google Scholar] [CrossRef] [PubMed]
- Kordis, D. A Genomic Perspective on the Chromodomain-Containing Retrotransposons: Chromoviruses. Gene 2005, 347, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Woudenberg, J.H.; Groenewald, J.Z.; Binder, M.; Crous, P.W. Alternaria Redefined. Stud. Mycol. 2013, 75, 171–212. [Google Scholar] [CrossRef] [PubMed]
- Selker, E.U. Premeiotic Instability of Repeated Sequences in Neurospora Crassa. Annu. Rev. Genet. 1990, 24, 579–613. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, A.S.; Vogan, A.A.; Gardiner, D.M.; Idnurm, A. Starships Are Active Eukaryotic Transposable Elements Mobilized by a New Family of Tyrosine Recombinases. Proc. Natl. Acad. Sci. USA 2023, 120, e2214521120. [Google Scholar] [CrossRef] [PubMed]
- Ciuffetti, L.M.; Manning, V.A.; Pandelova, I.; Betts, M.F.; Martinez, J.P. Host-Selective Toxins, Ptr ToxA and Ptr ToxB, as Necrotrophic Effectors in the Pyrenophora Tritici-Repentis-Wheat Interaction. New Phytol. 2010, 187, 911–919. [Google Scholar] [CrossRef]
- Stukenbrock, E.H.; McDonald, B.A. Geographical Variation and Positive Diversifying Selection in the Host-Specific Toxin SnToxA. Mol. Plant Pathol. 2007, 8, 321–332. [Google Scholar] [CrossRef]
- Singh, B.K.; Delgado-Baquerizo, M.; Egidi, E.; Guirado, E.; Leach, J.E.; Liu, H.; Trivedi, P. Climate Change Impacts on Plant Pathogens, Food Security and Paths Forward. Nat. Rev. Microbiol. 2023, 21, 640–656. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).