
Characterization and Optimization of Cellulose-Degrading Bacteria Isolated from Fecal Samples of Elaphurus davidianus Through Response Surface Methodology


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. Metagenomics Assembly and Gene Prediction
2.4. Isolation and Identification of Cellulose-Degrading Bacteria
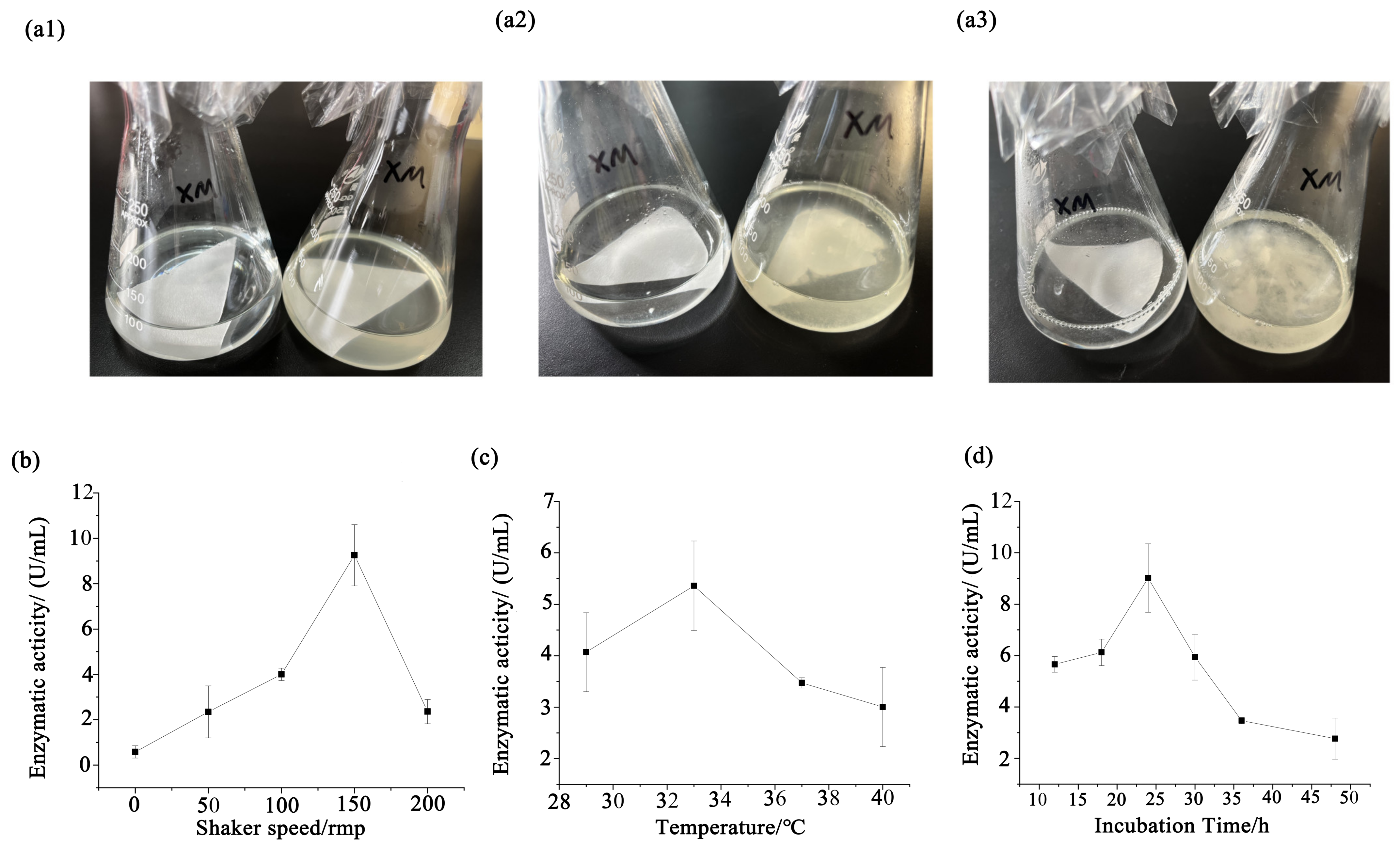
2.5. Cellulase Activity of Isolated Strain
2.6. Response Surface Curve Analysis
3. Results
3.1. Metagenomic Sequencing Results
3.1.1. Host Sequences and Gene Prediction
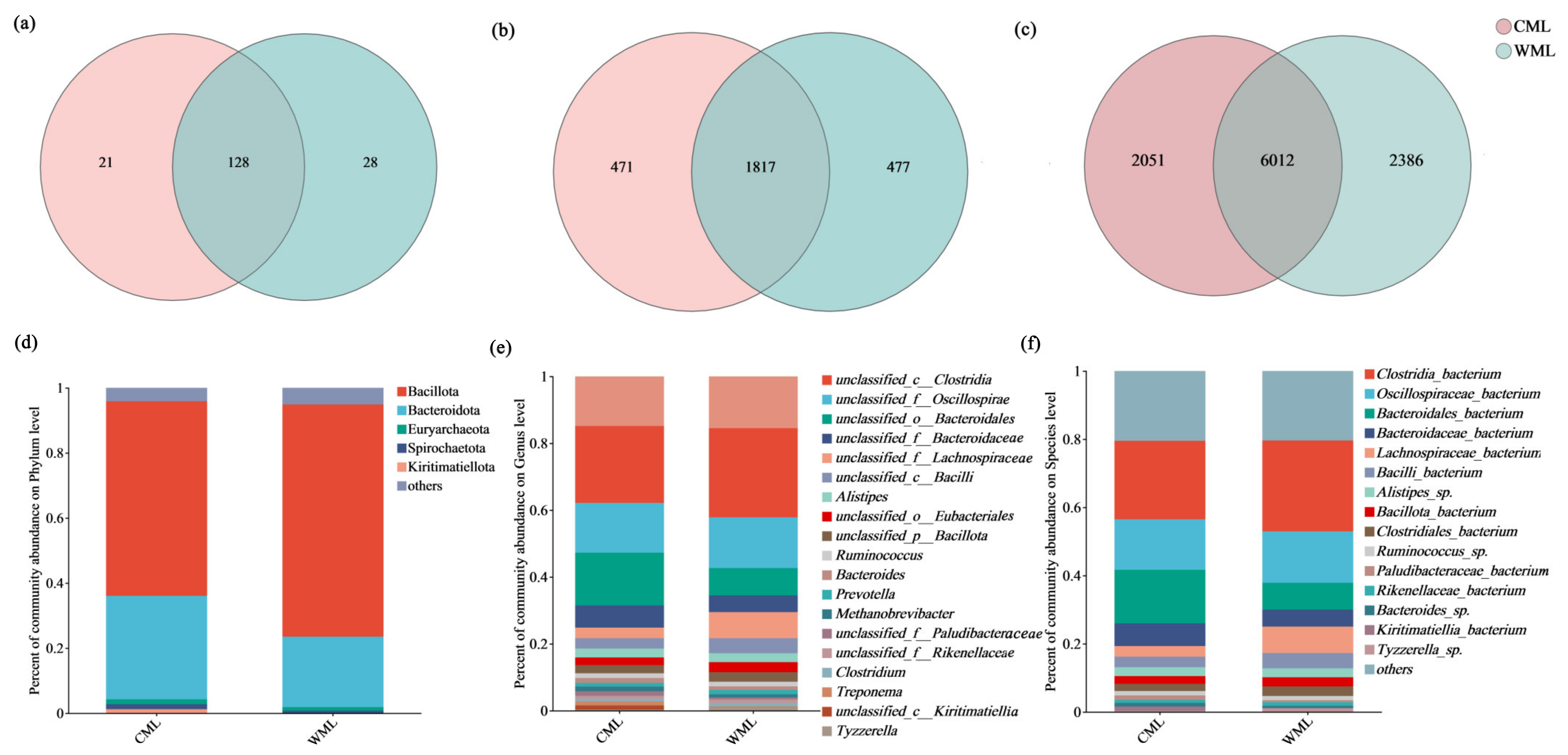
3.1.2. The Composition of Gut Microbiome
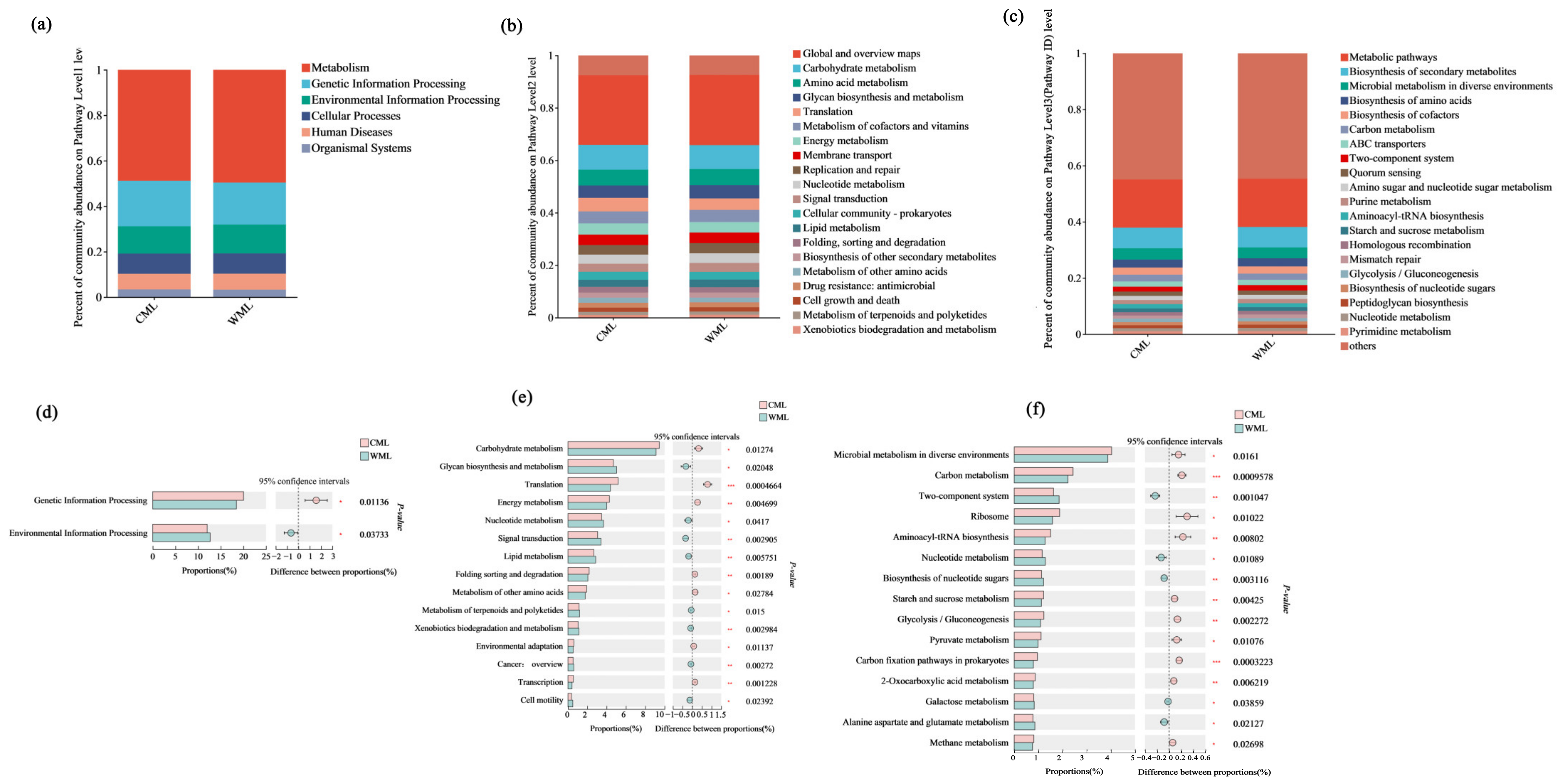
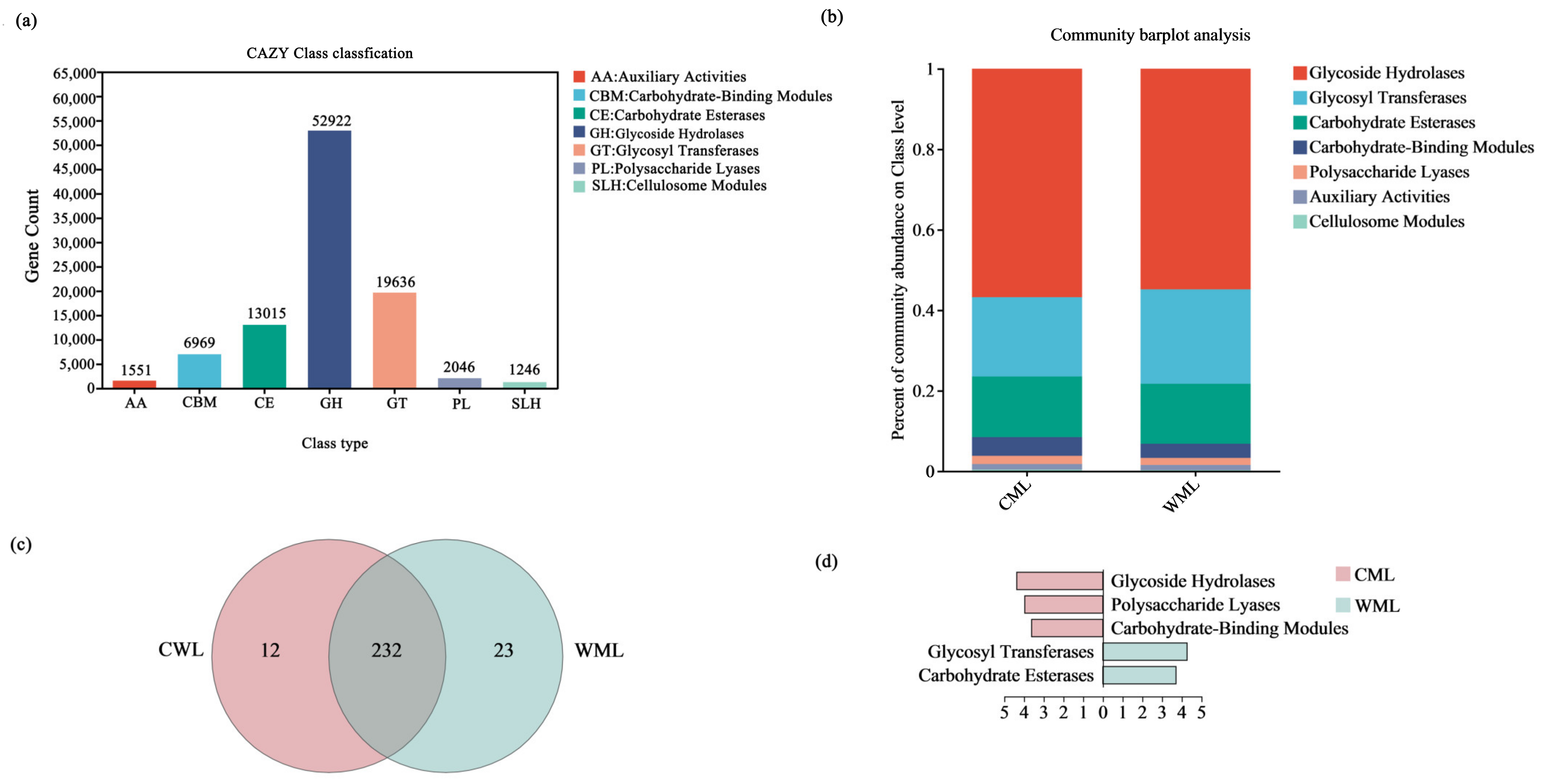
3.1.3. Comparison of Differences in Metabolic Levels Between Two Groups
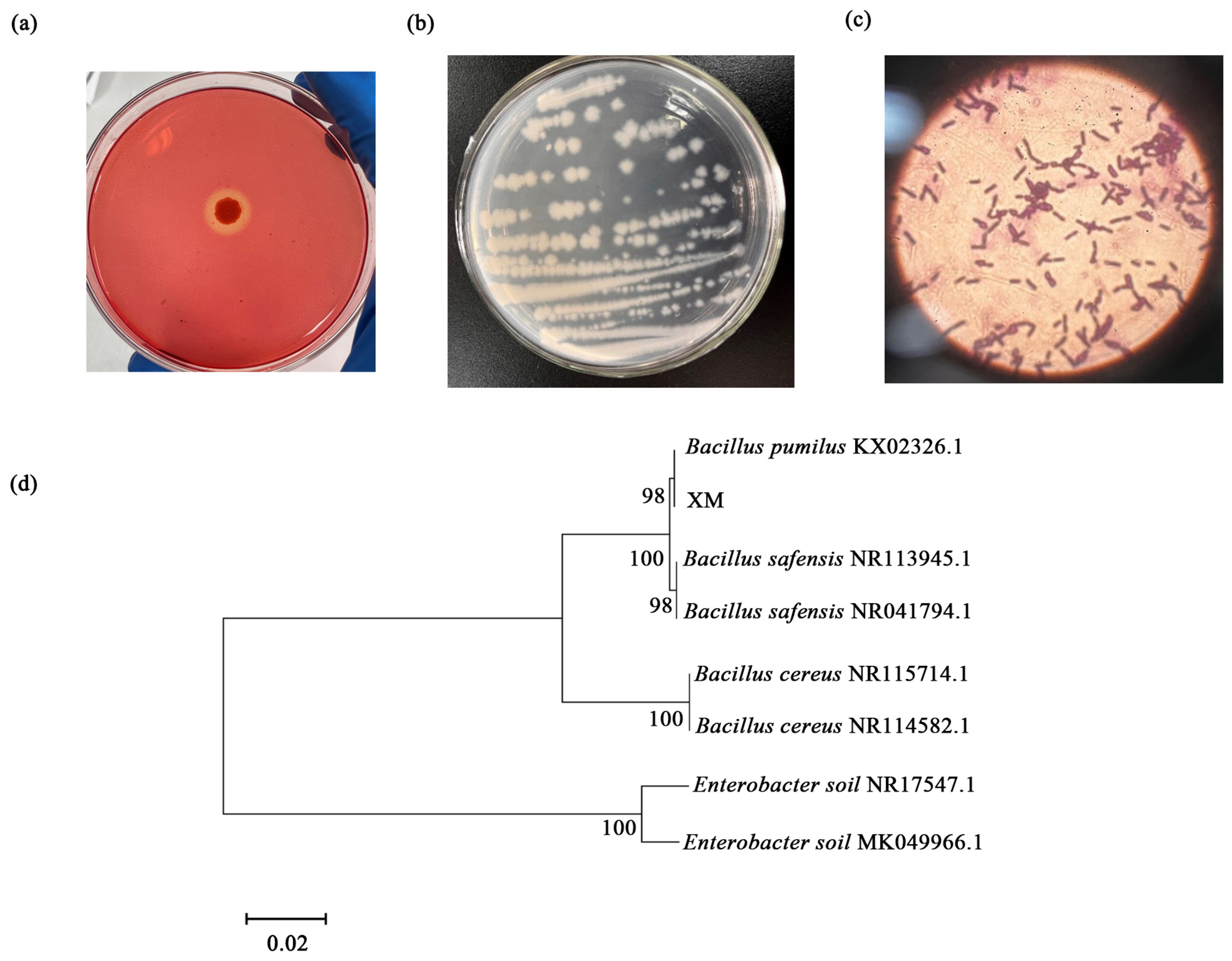
3.2. Isolation and Identification of Cellulolytic Bacteria
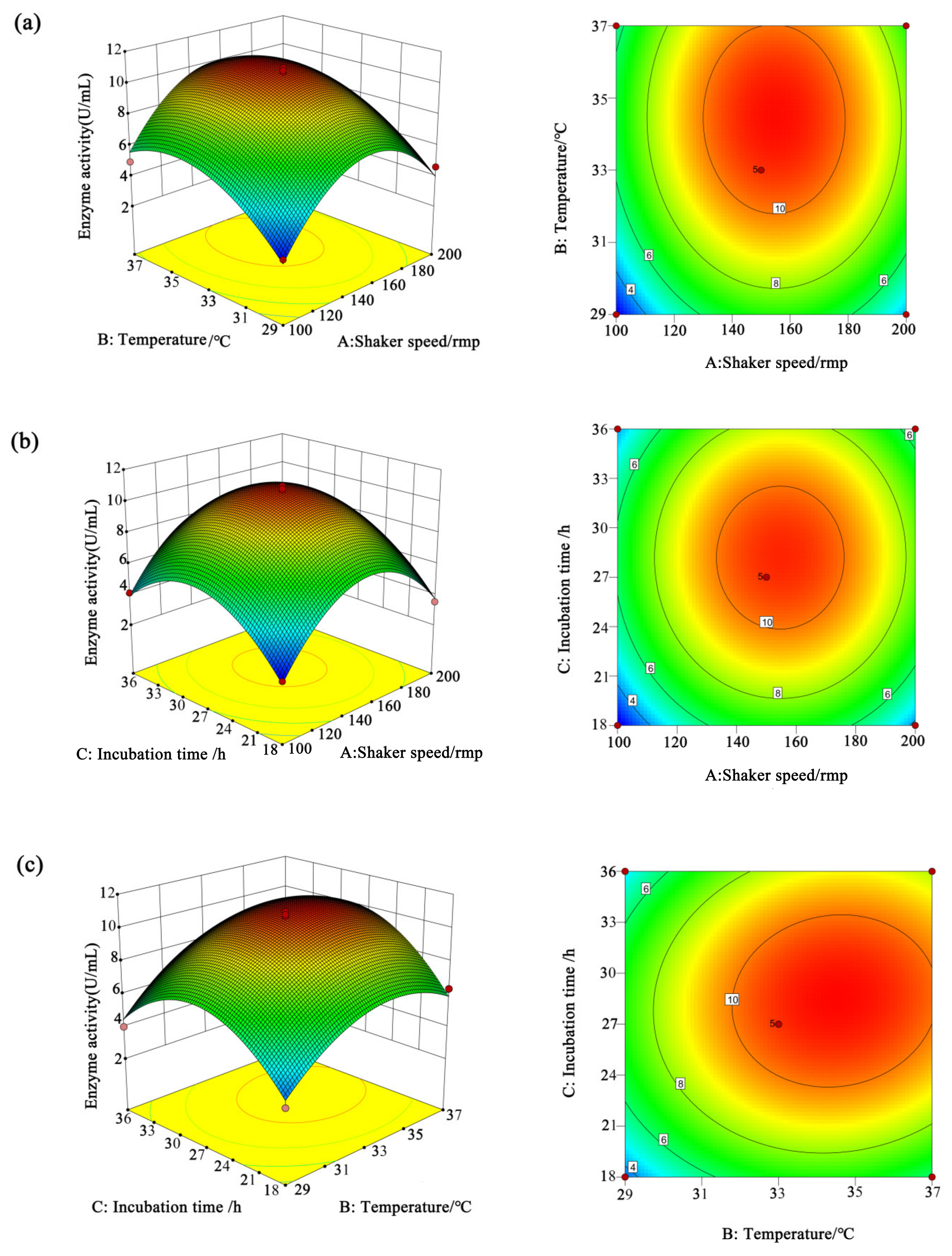
3.3. Statistical Optimization by Response Surface Methodology
3.3.1. Box–Behnken Design
3.3.2. The Results of CMCase by RSM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, W.; Li, X.H.; Zhang, Y.T. Research progress and the development trend of the utilization of crop straw biomass resources in China. Front. Chem. 2022, 10, 904660. [Google Scholar] [CrossRef] [PubMed]
- Lynd, L.R.; Weimer, P.J.; van Zyl, W.H.; Pretorius, I.S. Microbial cellulose utilization: Fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506–577. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.K.; Chatterjee, S.; Mazumder, P.B.; Gupta, S.K.; Sharma, S.; Vairale, M.G.; Datta, S.; Dwivedi, S.K.; Gupta, D.K. Bioethanol production from waste lignocelluloses: A review on microbial degradation potential. Chemosphere 2019, 231, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Ortega, M.; French, C.E. Engineering of industrially important microorganisms for assimilation of cellulosic biomass: Towards consolidated bioprocessing. Biochem. Soc. Trans. 2019, 47, 1781–1794. [Google Scholar] [CrossRef]
- Lai, D.M.; Deng, L.; Li, J.; Liao, B.; Guo, Q.X.; Fu, Y. Hydrolysis of cellulose into glucose by magnetic solid acid. Chemsuschem 2011, 4, 55–58. [Google Scholar] [CrossRef]
- Rubin, E.M. Genomics of cellulosic biofuels. Nature 2008, 454, 841–845. [Google Scholar] [CrossRef]
- Paul, M.; Mohapatra, S.; Kumar Das Mohapatra, P.; Thatoi, H. Microbial cellulases—An update towards its surface chemistry, genetic engineering and recovery for its biotechnological potential. Bioresour. Technol. 2021, 340, 125710. [Google Scholar] [CrossRef]
- Singhania, R.R.; Sukumaran, R.K.; Patel, A.K.; Larroche, C.; Pandey, A. Advancement and comparative profiles in the production technologies using solid-state and submerged fermentation for microbial cellulases. Enzym. Microb. Technol. 2010, 46, 541–549. [Google Scholar] [CrossRef]
- Yoon, L.W.; Ang, T.N.; Ngoh, G.C.; Chua, A.S.M. Fungal solid-state fermentation and various methods of enhancement in cellulase production. Biomass Bioenergy 2014, 67, 319–338. [Google Scholar] [CrossRef]
- Bajaj, P.; Mahajan, R. Cellulase and xylanase synergism in industrial biotechnology. Appl. Microbiol. Biotechnol. 2019, 103, 8711–8724. [Google Scholar] [CrossRef]
- Abou-Zeid, A.Z.; El-Diwany, A.I. Cellulose-decomposing fungi. Zentralbl. Bakteriol. Naturwiss 1978, 133, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, E.; Christakopoulos, P.; Katapodis, P.; Alexiou, A.; Vlachou, S.; Kekos, D.; Macris, B.J. Production and characterization of cellulolytic enzymes from the thermophilic fungus under solid state cultivation of agricultural wastes. Process. Biochem. 2003, 38, 1099–1104. [Google Scholar] [CrossRef]
- Peterson, R.; Nevalainen, H. Trichoderma reesei RUT-C30—Thirty years of strain improvement. Microbiology 2012, 158, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Boondaeng, A.; Keabpimai, J.; Trakunjae, C.; Vaithanomsat, P.; Srichola, P.; Niyomvong, N. Cellulase production under solid-state fermentation by Aspergillus sp. IN5: Parameter optimization and application. Heliyon 2024, 10, e26601. [Google Scholar] [CrossRef]
- Harnvoravongchai, P.; Singwisut, R.; Ounjai, P.; Aroonnual, A.; Kosiyachinda, P.; Janvilisri, T.; Chankhamhaengdecha, S. Isolation and characterization of thermophilic cellulose and hemicellulose degrading bacterium, Thermoanaerobacterium sp. R63 from tropical dry deciduous forest soil. PLoS ONE 2020, 15, e0236518. [Google Scholar] [CrossRef]
- Du, C.J.; Li, C.X.; Cao, P.; Li, T.T.; Du, D.D.; Wang, X.J.; Zhao, J.W.; Xiang, W.S. Massilia cellulosiltytica sp. nov., a novel cellulose-degrading bacterium isolated from rhizosphere soil of rice (Oryza sativa L.) and its whole genome analysis. Antonie Van Leeuwenhoek 2021, 114, 1529–1540. [Google Scholar] [CrossRef]
- He, Z.; Ding, B.P.; Ali, Q.; Liu, H.Y.; Zhao, Y.; Wang, X.J.; Han, Y.Z.; Dong, H.; Divvela, P.K.; Juan, Y.H. Screening and isolation of cold-adapted cellulose degrading bacterium: A candidate for straw degradation and De novo genome sequencing analysis. Front. Microbiol. 2022, 13, 1098723. [Google Scholar] [CrossRef]
- Li, H.; Zhang, M.Q.; Zhang, Y.H.; Xu, X.M.; Zhao, Y.; Jiang, X.P.; Zhang, R.; Gui, Z.Z. Characterization of cellulose-degrading bacteria isolated from silkworm excrement and optimization of its cellulase production. Polymers 2023, 15, 4142. [Google Scholar] [CrossRef]
- Manzoor, N.; Gao, L.B.; Deng, D.D.; Liu, Z.Z.; Jiang, Y.J. Cellulase extraction from cellulolytic bacteria promoting bioelectricity production by degrading cellulose. J. Electroanal. Chem. 2018, 829, 241–248. [Google Scholar] [CrossRef]
- Singh, S.; Thavamani, P.; Megharaj, M.; Naidu, R. Multifarious activities of cellulose degrading bacteria from Koala (Phascolarctos cinereus) faeces. J. Anim. Sci. Technol. 2015, 57, 23. [Google Scholar] [CrossRef]
- Ungerfeld, E.M.; Leigh, M.B.; Forster, R.J.; Barboza, P.S. Influence of season and diet on fiber digestion and bacterial community structure in the rumen of Muskoxen (Ovibos moschatus). Microorganisms 2018, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Ushakova, N.A.; Belov, L.P.; Varshavskiĭ, A.A.; Kozlova, A.A.; Kolganova, T.V.; Bulygina, E.S.; Turova, T.P. Cellulose decomposition under nitrogen deficiency by bacteria isolated from the intestines of phytophagous vertebrates. Microbiology 2003, 72, 356–362. [Google Scholar] [CrossRef]
- Wang, H.L.; Zhao, Y.; Wang, F.J.; Sun, X.J.; Zhu, J.Q.; Zhang, Y.M.; Wei, S.D.; Chen, H. Diet composition and selection of Pere David’s deer in Hubei Shishou Milu National Nature Reserve, China. Ecol. Evol. 2023, 13, e9702. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.H.; Liu, H.Y.; Liu, B.; Yuan, B.D.; Lu, C.H. Analysis of the gut microbiome of wild and captive Pere David’s Deer. Front. Microbiol. 2019, 10, 2331. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-Throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef]
- Yao, H.; Mo, Q.; Wu, H.; Zhao, D. How do living conditions affect the gut microbiota of endangered Père David’s deer (Elaphurus davidianus)? Peer J. 2023, 11, e14897. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Noguchi, H.; Park, J.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Li, D.H.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Fu, L.M.; Niu, B.F.; Zhu, Z.W.; Wu, S.T.; Li, W.Z. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Drula, E.; Garron, M.L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Darwesh, O.M.; El-Maraghy, S.H.; Abdel-Rahman, H.M.; Zaghloul, R.A. Improvement of paper wastes conversion to bioethanol using novel cellulose degrading fungal isolate. Fuel 2020, 262, 116581. [Google Scholar] [CrossRef]
- Zhou, J.; Yin, L.H.; Wu, C.B.; Wu, S.J.; Lu, J.D.; Fang, H.L.; Qian, Y.C. Screening of an alkaline CMCase-producing strain and the optimization of its fermentation condition. Curr. Pharm. Biotechnol. 2020, 21, 1304–1315. [Google Scholar] [CrossRef]
- Kognou, A.L.M.; Chio, C.L.; Khatiwada, J.R.; Shrestha, S.; Chen, X.T.; Han, S.H.; Li, H.W.; Jiang, Z.H.; Xu, C.C.; Qin, W.S. Characterization of cellulose-degrading bacteria isolated from soil and the optimization of their culture conditions for cellulase production. Appl. Biochem. Biotechnol. 2022, 194, 5060–5082. [Google Scholar] [CrossRef]
- Yadav, S.; Dubey, S.K. Cellulose degradation potential of strain BHU3 and its whole genome sequence. Bioresour. Technol. 2018, 262, 124–131. [Google Scholar] [CrossRef]
- Tang, H.; Li, Y.Q.; Zheng, L.; Wang, M.J.; Luo, C.B. Efficient saccharification of bamboo biomass by secretome protein of the cellulolytic bacterium LY1 based on whole-genome and secretome analysis. Renew. Energy 2022, 193, 32–40. [Google Scholar] [CrossRef]
- Anand, A.A.P.; Vennison, S.J.; Sankar, S.G.; Prabhu, D.I.; Vasan, P.T.; Raghuraman, T.; Geoffrey, C.J.; Vendan, S.E. Isolation and characterization of bacteria from the gut of that degrade cellulose, xylan, pectin and starch and their impact on digestion. J. Insect Sci. 2010, 10, 107. [Google Scholar] [CrossRef]
- Kumawat, P.K.; Sahoo, A.; Sarkar, S.; Kumar, S. Isolation and characterization of anaerobic bacteria with fiber degradation potential from faeces of grazing on semi arid Indian conditions. Arch. Microbiol. 2021, 203, 5105–5116. [Google Scholar] [CrossRef] [PubMed]
- Gullert, S.; Fischer, M.A.; Turaev, D.; Noebauer, B.; Ilmberger, N.; Wemheuer, B.; Alawi, M.; Rattei, T.; Daniel, R.; Schmitz, R.A.; et al. Deep metagenome and metatranscriptome analyses of microbial communities affiliated with an industrial biogas fermenter, a cow rumen, and elephant feces reveal major differences in carbohydrate hydrolysis strategies. Biotechnol. Biofuels 2016, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhan, M.Y.; Pei, E. Succession of intestinal microbial structure of Giant Pandas (Ailuropoda melanoleuca) during different developmental stages and its correlation with cellulase activity. Animals 2021, 11, 2358. [Google Scholar] [CrossRef]
- Guo, W.; Mishra, S.; Wang, C.D.; Zhang, H.M.; Ning, R.H.; Kong, F.; Zeng, B.; Zhao, J.C.; Li, Y. Comparative study of gut microbiota in wild and captive Giant Pandas (Ailuropoda melanoleuca). Genes 2019, 10, 827. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Zhang, L.Z.; Fan, C.; Liu, C.F.; Li, W.J.; Li, J.Y.; Zhao, X.Q.; Jia, S.G.; Zhang, Y.M. Domestication shapes the community structure and functional metagenomic content of the yak fecal microbiota. Front. Microbiol. 2021, 12, 594075. [Google Scholar] [CrossRef]
- Lapidus, A.L.; Korobeynikov, A.I. Metagenomic Data Assembly—The way of decoding unknown microorganisms. Front Microbiol. 2021, 12, 613791. [Google Scholar] [CrossRef]
- Ghurye, J.S.; Cepeda-Espinoza, V.; Pop, M. Metagenomic assembly: Overview, challenges and applications. Yale J. Biol. Med. 2016, 89, 353–362. [Google Scholar]
- Liang, J.; Fang, W.; Wang, Q.; Zubair, M.; Zhang, G.; Ma, W.; Cai, Y.; Zhang, P. Metagenomic analysis of community, enzymes and metabolic pathways during corn straw fermentation with rumen microorganisms for volatile fatty acid production. Bioresour. Technol. 2021, 342, 126004. [Google Scholar] [CrossRef]
- Liang, J.; Chang, J.; Zhang, R.; Fang, W.; Chen, L.; Ma, W.; Zhang, Y.; Yang, W.; Li, Y.; Zhang, P.; et al. Metagenomic analysis reveals the efficient digestion mechanism of corn stover in Angus bull rumen: Microbial community succession, CAZyme composition and functional gene expression. Chemosphere 2023, 336, 139242. [Google Scholar] [CrossRef]
- Jegousse, C.; Vannier, P.; Groben, R.; Glöckner, F.O.; Marteinsson, V. A total of 219 metagenome-assembled genomes of microorganisms from Icelandic marine waters. Peer J. 2021, 9, e11112. [Google Scholar] [CrossRef]
- Tyson, G.W.; Lo, I.; Baker, B.J.; Allen, E.E.; Hugenholtz, P.; Banfield, J.F. Genome-directed isolation of the key nitrogen fixer Leptospirillum ferrodiazotrophum sp. nov. from an acidophilic microbial community. Appl. Environ. Microbiol. 2005, 71, 6319–6324. [Google Scholar] [CrossRef] [PubMed]
- Pope, P.B.; Smith, W.; Denman, S.E.; Tringe, S.G.; Barry, K.; Hugenholtz, P.; McSweeney, C.S.; McHardy, A.C.; Morrison, M. Isolation of succinivibrionaceae implicated in low methane emissions from Tammar Wallabies. Science 2011, 333, 646–648. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Duranti, S.; Alessandri, G.; Turroni, F.; Mancabelli, L.; Tatoni, D.; Ossiprandi, M.C.; van Sinderen, D.; Ventura, M. Isolation of novel gut bifidobacteria using a combination of metagenomic and cultivation approaches. Genome Biol. 2019, 20, 96. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Xu, L.; Lu, G.; Zhu, L. Evaluation of the function of wild animal gut microbiomes using next-generation sequencing and bioinformatics and its relevance to animal conservation. Evol. Bioinform. Online 2019, 15, 1176934319848438. [Google Scholar] [CrossRef]
- Yang, W.P.; Meng, F.; Peng, J.; Han, P.; Fang, F.; Ma, L.; Cao, B. Isolation and identification of a cellulolytic bacterium from the Tibetan pig’s intestine and investigation of its cellulase production. Electron J. Biotechnol. 2014, 17, 262–267. [Google Scholar] [CrossRef]
- Lugani, Y.; Singla, R.; Sooch, B.S. Optimization of cellulase production from newly isolated Bacillus sp. Y3. J. Bioprocess Biotech. 2015, 5, 11. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | Numnber | Level | ||
|---|---|---|---|---|
| −1 | 0 | 1 | ||
| Shaker speed/rmp | A | 100 | 150 | 200 |
| Temperature/°C | B | 29 | 33 | 37 |
| Incubation time/h | C | 18 | 27 | 36 |
| Number | A | B | C | Enzymatic Activity U/mL |
|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 10.32 |
| 2 | −1 | 0 | 1 | 4.13 |
| 3 | 1 | 0 | 1 | 5.19 |
| 4 | 1 | −1 | 0 | 4.62 |
| 5 | 0 | −1 | 1 | 4 |
| 6 | 0 | 0 | 0 | 10.68 |
| 7 | 0 | 0 | 0 | 10.26 |
| 8 | −1 | −1 | 0 | 2.71 |
| 9 | 1 | 0 | −1 | 3.54 |
| 10 | 0 | −1 | −1 | 3.13 |
| 11 | 0 | 0 | 0 | 10.81 |
| 12 | 1 | 1 | 0 | 6.6 |
| 13 | 0 | 0 | 0 | 10.98 |
| 14 | 0 | 1 | −1 | 6.36 |
| 15 | 0 | 1 | 1 | 8.55 |
| 16 | −1 | 1 | 0 | 4.95 |
| 17 | −1 | 0 | −1 | 2.54 |
| Source | Sum of Squares | df | Mean Square | F Value | p Value | ||
|---|---|---|---|---|---|---|---|
| Model | 156.43 | 9 | 17.38 | 53.89 | <0.0001 | significant | |
| A | 3.95 | 1 | 3.95 | 12.24 | 0.0100 | ||
| B | 18.00 | 1 | 18.00 | 55.80 | 0.0001 | ||
| C | 4.96 | 1 | 4.96 | 15.38 | 0.0057 | ||
| AB | 0.017 | 1 | 0.017 | 0.052 | 0.8255 | ||
| AC | 0.0009 | 1 | 0.0009 | 0.00279 | 0.9593 | ||
| BC | 0.44 | 1 | 0.44 | 1.35 | 0.2833 | ||
| A2 | 60.00 | 1 | 60.00 | 186.02 | <0.0001 | ||
| B2 | 18.83 | 1 | 18.83 | 58.39 | 0.0001 | ||
| C2 | 37.52 | 1 | 37.52 | 116.31 | <0.0001 | ||
| Residual | 2.26 | 7 | 0.32 | ||||
| Lack of Fit | 1.87 | 3 | 0.62 | 6.42 | 0.0522 | not significant | |
| Pure Error | 0.39 | 4 | 0.097 | ||||
| Cor Total | 158.69 | 16 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.; Shi, C.; Xu, T.; Dai, X.; Zhao, D. Characterization and Optimization of Cellulose-Degrading Bacteria Isolated from Fecal Samples of Elaphurus davidianus Through Response Surface Methodology. Microorganisms 2025, 13, 348. https://doi.org/10.3390/microorganisms13020348
Wu H, Shi C, Xu T, Dai X, Zhao D. Characterization and Optimization of Cellulose-Degrading Bacteria Isolated from Fecal Samples of Elaphurus davidianus Through Response Surface Methodology. Microorganisms. 2025; 13(2):348. https://doi.org/10.3390/microorganisms13020348
Chicago/Turabian StyleWu, Hong, Chunmiao Shi, Tianyi Xu, Xinrui Dai, and Dapeng Zhao. 2025. "Characterization and Optimization of Cellulose-Degrading Bacteria Isolated from Fecal Samples of Elaphurus davidianus Through Response Surface Methodology" Microorganisms 13, no. 2: 348. https://doi.org/10.3390/microorganisms13020348
APA StyleWu, H., Shi, C., Xu, T., Dai, X., & Zhao, D. (2025). Characterization and Optimization of Cellulose-Degrading Bacteria Isolated from Fecal Samples of Elaphurus davidianus Through Response Surface Methodology. Microorganisms, 13(2), 348. https://doi.org/10.3390/microorganisms13020348