

γBMGC: A Comprehensive and Accurate Database for Screening TMAO-Associated Cardiovascular Diseases

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
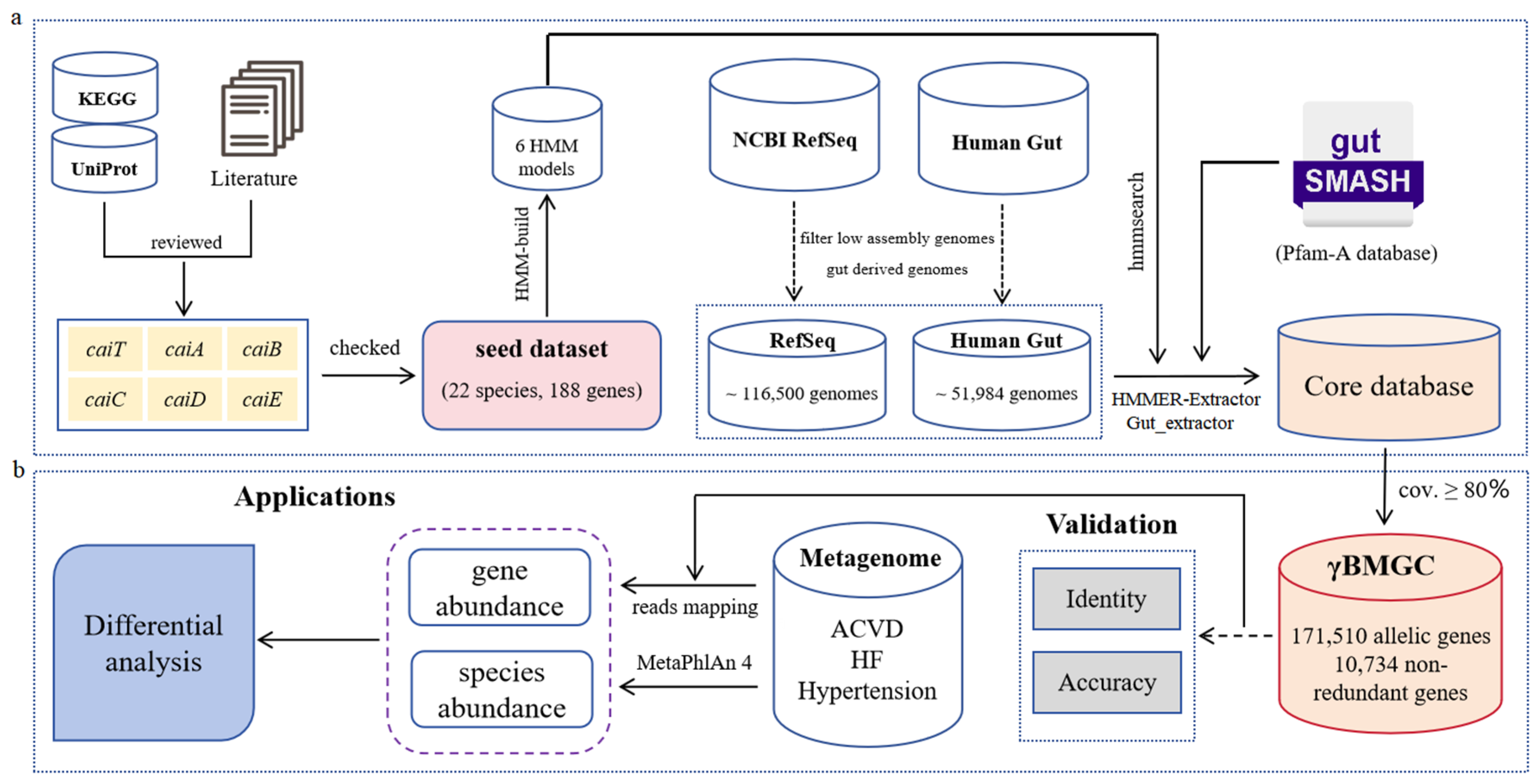
2.1. Construction of the γBMGC Database
2.2. Simulated Gene Datasets
2.3. Genome Sequence Datasets from a Mock Community
2.4. Metagenome Sequencing Datasets
2.5. Gene Annotation and Abundance Analysis
2.6. Statistical and Case Study
3. Results
3.1. Construction of the Seed Dataset
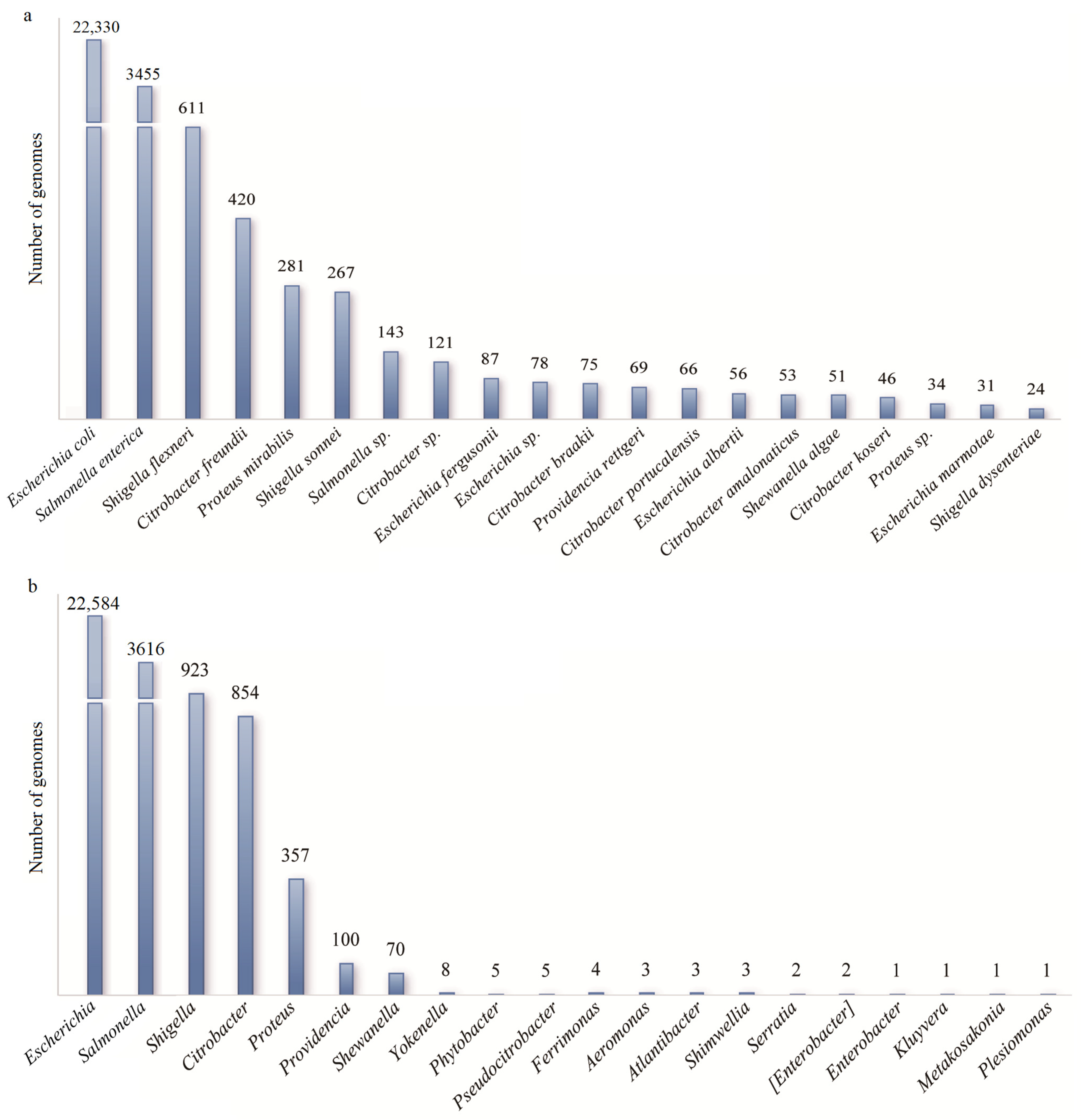
3.2. The Database of Metabolic Gene Clusters Associated with γ-Butylbetaine
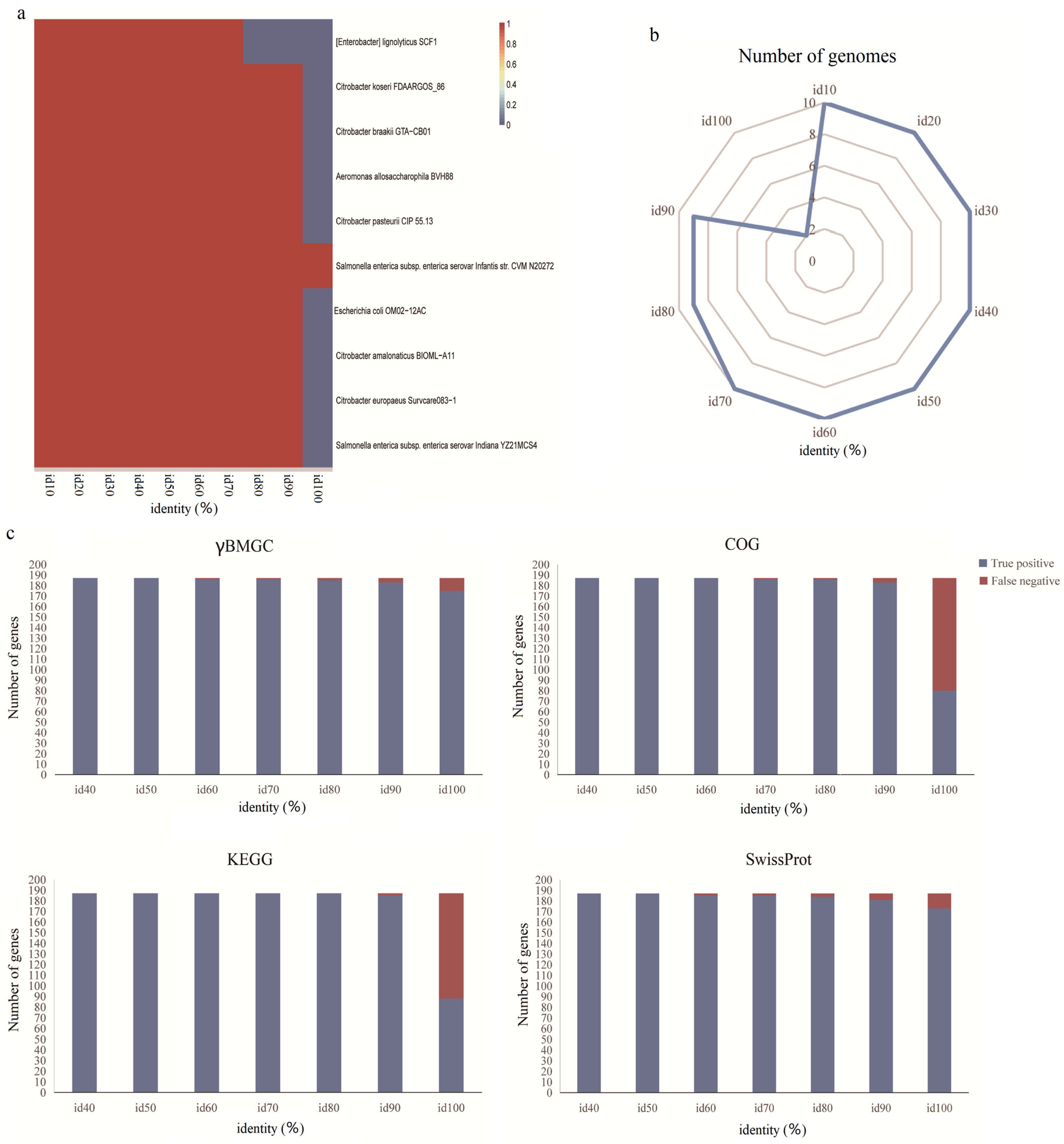
3.3. Benchmark of the γBMGC Database
3.4. Validation of γBMGC with a Mock Community
3.5. Performance Comparison Among Different Orthology Databases
3.6. Application of γBMGC in Analysis of Cardiovascular Diseases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buffa, J.A.; Romano, K.A.; Copeland, M.F.; Cody, D.B.; Zhu, W.; Galvez, R.; Fu, X.; Ward, K.; Ferrell, M.; Dai, H.J.; et al. The microbial gbu gene cluster links cardiovascular disease risk associated with red meat consumption to microbiota L-carnitine catabolism. Nat. Microbiol. 2022, 7, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Lam-Galvez, B.R.; Kirsop, J.; Wang, Z.; Levison, B.S.; Gu, X.; Copeland, M.F.; Bartlett, D.; Cody, D.B.; Dai, H.J.; et al. l-Carnitine in omnivorous diets induces an atherogenic gut microbial pathway in humans. J. Clin. Investig. 2019, 129, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Levison, B.S.; Culley, M.K.; Buffa, J.A.; Wang, Z.; Gregory, J.C.; Org, E.; Wu, Y.; Li, L.; Smith, J.D.; et al. Gamma-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab. 2014, 20, 799–812. [Google Scholar] [CrossRef]
- Skagen, K.; Troseid, M.; Ueland, T.; Holm, S.; Abbas, A.; Gregersen, I.; Kummen, M.; Bjerkeli, V.; Reier-Nilsen, F.; Russell, D.; et al. The Carnitine-butyrobetaine-trimethylamine-N-oxide pathway and its association with cardiovascular mortality in patients with carotid atherosclerosis. Atherosclerosis 2016, 247, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Eichler, K.; Bourgis, F.; Buchet, A.; Kleber, H.; Mandrand-Berthelot, M. Molecular characterization of the cai operon necessary for carnitine metabolism in Escherichia coli. Mol. Microbiol. 1994, 13, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Rebouche, C.J. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann. N. Y. Acad. Sci. 2004, 1033, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Jameson, E.; Quareshy, M.; Chen, Y. Methodological considerations for the identification of choline and carnitine-degrading bacteria in the gut. Methods 2018, 149, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Bernal, V.; Masdemont, B.; Arense, P.; Cánovas, M.; Iborra, J.L. Redirecting metabolic fluxes through cofactor engineering: Role of CoA-esters pool during L-carnitine production by Escherichia coli. J. Biotechnol. 2007, 132, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Zeng, J.; Tu, Q.; Yu, X.; Qian, L.; Wang, C.; Shu, L.; Liu, F.; Liu, S.; Huang, Z.; He, J.; et al. PCycDB: A comprehensive and accurate database for fast analysis of phosphorus cycling genes. Microbiome 2022, 10, 101. [Google Scholar] [CrossRef]
- Yang, J.; Sun, S.; Sun, N.; Lu, L.; Zhang, C.; Shi, W.; Zhao, Y.; Jia, S. HMMER-Extractor: An auxiliary toolkit for identifying genomic macromolecular metabolites based on Hidden Markov Models. Int. J. Biol. Macromol. 2024, 283 Pt 2, 137666. [Google Scholar] [CrossRef]
- Pascal, A.V.; Roel-Touris, J.; Dodd, D.; Fischbach, M.A.; Medema, M.H. The gutSMASH web server: Automated identification of primary metabolic gene clusters from the gut microbiota. Nucleic Acids Res. 2021, 49, W263–W270. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Ye, L.; Li, J.; Wang, W.; Li, S.; Bao, M.; Wu, S.; Li, L.; Geng, B.; Zhou, X.; et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci. Rep. 2018, 8, 635. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef]
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files, Version 1.33. 2011. Available online: https://github.com/najoshi/sickle (accessed on 2 March 2024).
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP-A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Oberg, N.; Zallot, R.; Gerlt, J.A. EFI-EST, EFI-GNT, and EFI-CGFP: Enzyme function initiative (EFI) web resource for genomic enzymology tools. J. Mol. Biol. 2023, 435, 168018. [Google Scholar] [CrossRef]
- Zallot, R.; Oberg, N.; Gerlt, J.A. The EFI web resource for genomic enzymology tools: Leveraging protein, genome, and metagenome databases to discover novel enzymes and metabolic pathways. Biochemistry 2019, 58, 4169–4182. [Google Scholar] [CrossRef] [PubMed]
- Manghi, P.; Blanco-Míguez, A.; Manara, S.; NabiNejad, A.; Cumbo, F.; Beghini, F.; Armanini, F.; Golzato, D.; Huang, K.D.; Thomas, A.M.; et al. MetaPhlAn 4 profiling of unknown species-level genome bins improves the characterization of diet-associated microbiome changes in mice. Cell Rep. 2023, 42, 112464. [Google Scholar] [CrossRef]
- Yu, X.L.; Zhou, J.Y.; Song, W.; Xu, M.; He, Q.; Peng, Y.; Tian, Y.; Wang, C.; Shu, L.; Wang, S.; et al. SCycDB: A curated functional gene database for metagenomic profiling of sulphur cycling pathways. Environ. Microbiol. 2021, 21, 924–940. [Google Scholar] [CrossRef]
- Zhang, A.N.; Li, L.G.; Ma, L.; Gillings, M.R.; Tiedje, J.M.; Zhang, T. Conserved phylogenetic distribution and limited antibiotic resistance of class 1 integrons revealed by assessing the bacterial genome and plasmid collection. Microbiome 2018, 6, 130. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; A Wlodarski, M.; Edalatmand, A.; Petkau, A.; A Syed, S.; Tsang, K.K.; et al. CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Lin, L.; Cheng, L.; Deng, Y.; He, Z. NCycDB: A curated integrative database for fast and accurate metagenomic profiling of nitrogen cycling genes. Bioinformatics 2019, 35, 1040–1048. [Google Scholar] [CrossRef]
- Arango-Argoty, G.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Ueland, T.; Svardal, A.; Oie, E.; Askevold, E.T.; Nymoen, S.H.; Bjørndal, B.; Dahl, C.P.; Gullestad, L.; Berge, R.K.; Aukrust, P. Disturbed carnitine regulation in chronic heart failure—Increased plasma levels of palmitoyl-carnitine are associated with poor prognosis. Int. J. Cardiol. 2013, 167, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Heinrich-Sanchez, Y. A small, polyphyletic group of Firmicutes synthesizes trimethylamine from l-carnitine. mLife 2023, 2, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Trøseid, M.; Mayerhofer, C.C.; Broch, K.; Arora, S.; Svardal, A.; Hov, J.R.; Andreassen, A.K.; Gude, E.; Karason, K.; Dellgren, G.; et al. The carnitine-butyrobetaine-TMAO pathway after cardiac transplant: Impact on cardiac allograft vasculopathy and acute rejection. J. Heart Lung Transplant. 2019, 38, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Tao, T.; Yu, G.; Zhang, H.; Wu, Y.; Sun, S.; Guo, K.; Jia, S. γBMGC: A Comprehensive and Accurate Database for Screening TMAO-Associated Cardiovascular Diseases. Microorganisms 2025, 13, 225. https://doi.org/10.3390/microorganisms13020225
Yang G, Tao T, Yu G, Zhang H, Wu Y, Sun S, Guo K, Jia S. γBMGC: A Comprehensive and Accurate Database for Screening TMAO-Associated Cardiovascular Diseases. Microorganisms. 2025; 13(2):225. https://doi.org/10.3390/microorganisms13020225
Chicago/Turabian StyleYang, Guang, Tiantian Tao, Guohao Yu, Hongqian Zhang, Yiwen Wu, Siqi Sun, Kexin Guo, and Shulei Jia. 2025. "γBMGC: A Comprehensive and Accurate Database for Screening TMAO-Associated Cardiovascular Diseases" Microorganisms 13, no. 2: 225. https://doi.org/10.3390/microorganisms13020225
APA StyleYang, G., Tao, T., Yu, G., Zhang, H., Wu, Y., Sun, S., Guo, K., & Jia, S. (2025). γBMGC: A Comprehensive and Accurate Database for Screening TMAO-Associated Cardiovascular Diseases. Microorganisms, 13(2), 225. https://doi.org/10.3390/microorganisms13020225