
Comparative Genomics Reveals Evidence of the Genome Reduction and Metabolic Potentials of Aliineobacillus hadale Isolated from Challenger Deep Sediment of the Mariana Trench

Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Description
2.2. Bacterial Isolation
2.3. Microbial Utilization of the Substrates and Chemotaxonomy
2.4. Genomic DNA Extraction, Sequencing, and Assembly
2.5. 16S rRNA Prediction and Comparison
2.6. Gene Annotation and Genomic Comparison
2.7. Phylogenetic Analysis
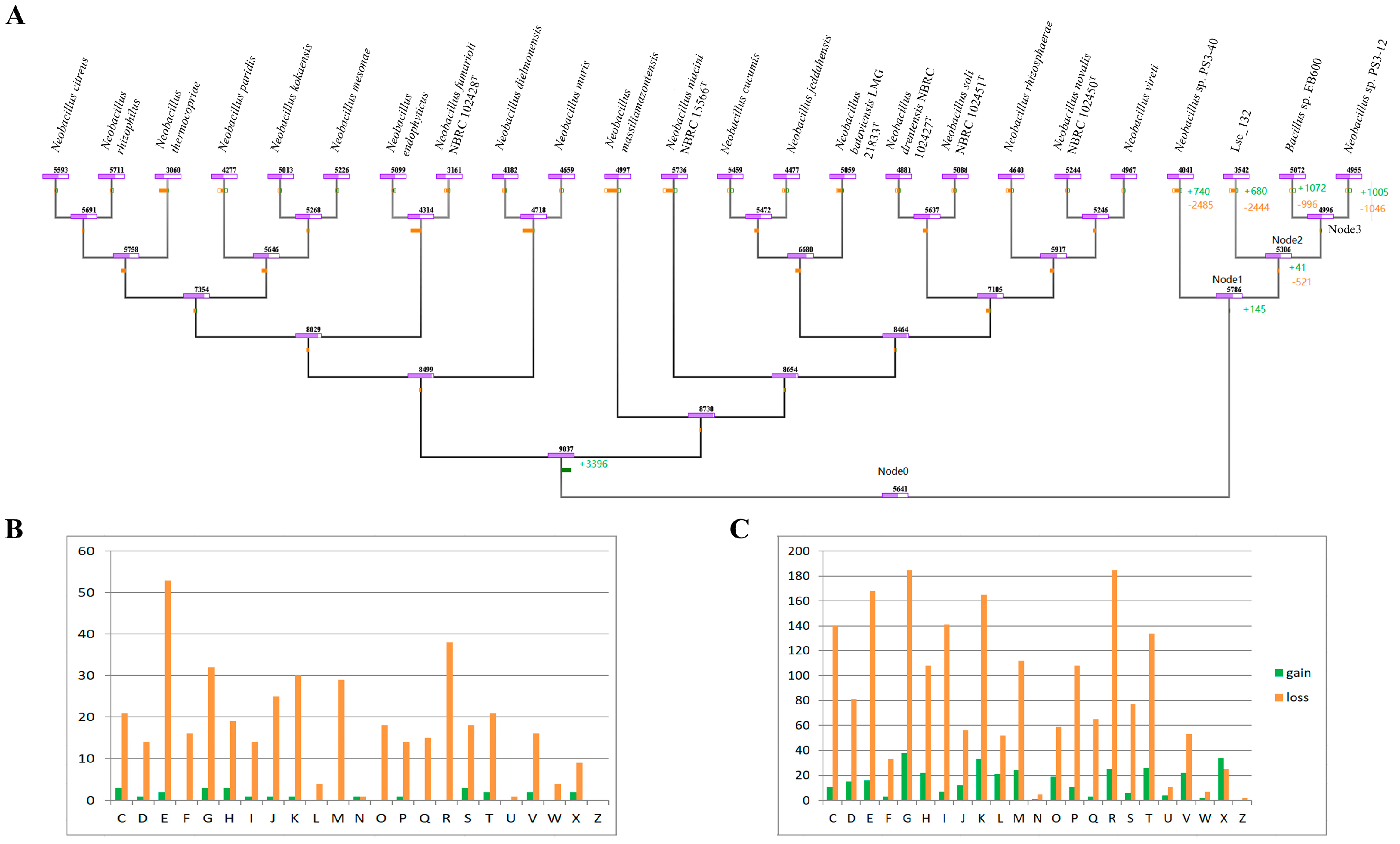
2.8. Gene Gain and Loss Analysis
3. Results and Discussion
3.1. Description of Strain Lsc_1132T
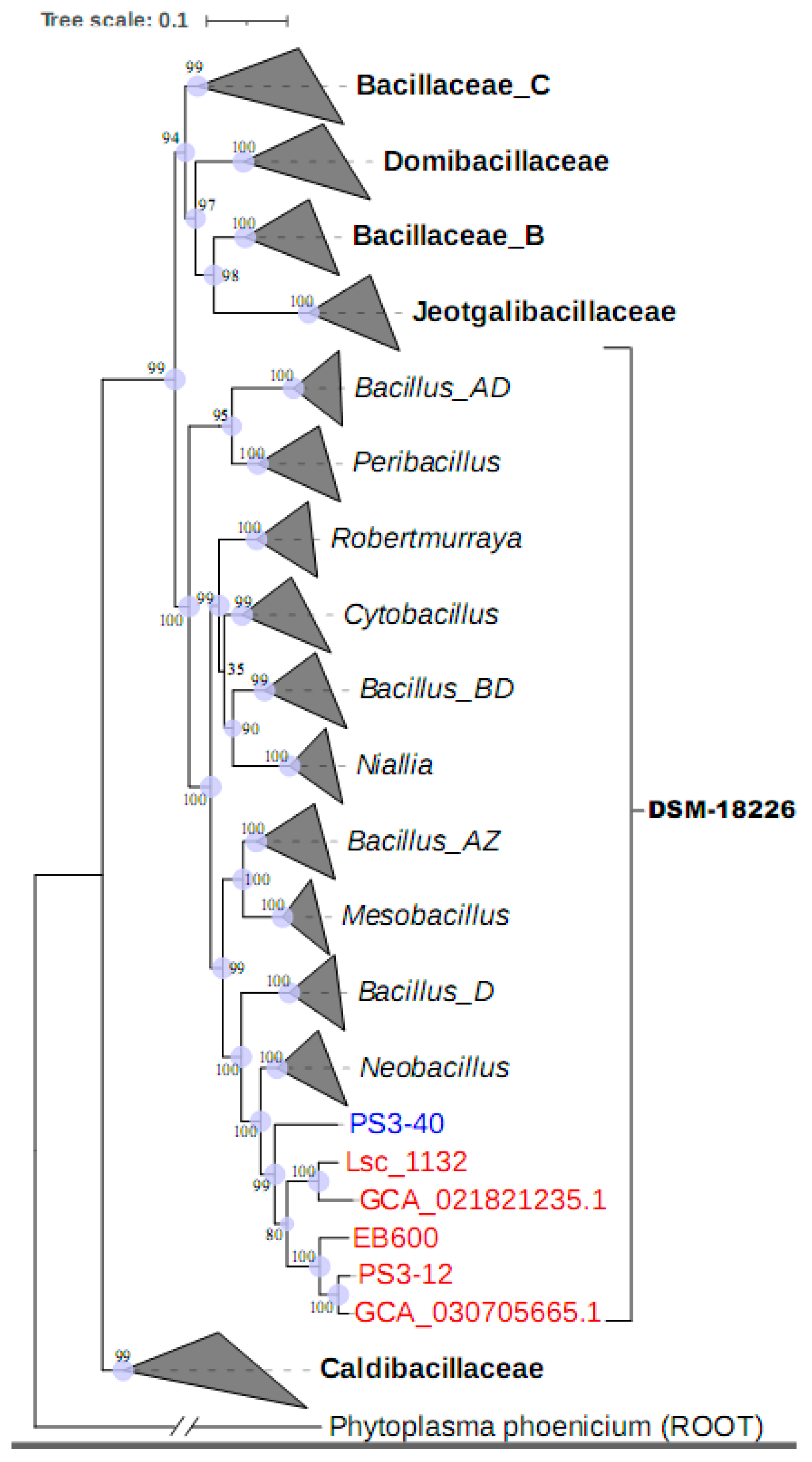
3.2. The Phylogenetic Characteristics of Strain Lsc_1132T
3.3. Description of Genomic Features
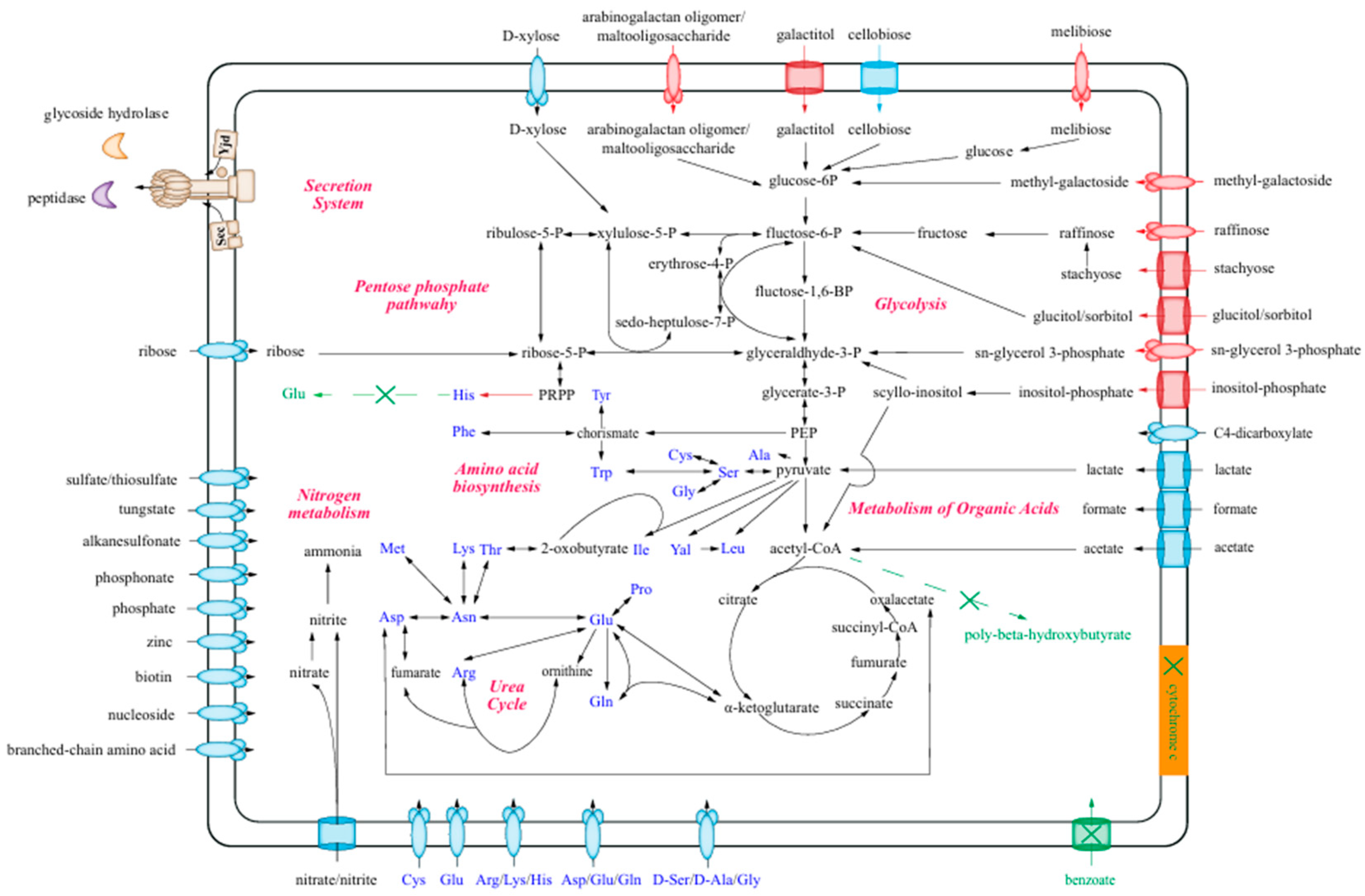
3.4. The Metabolic Characteristics of Strain Lsc_1132T
3.4.1. The Utilization of Carbohydrates
3.4.2. Metabolisms of Amino Acids
3.4.3. Metabolisms of Organic Acids
3.4.4. Nitrogen Metabolism
4. Discussion
5. Conclusions
5.1. Description of Aliineobacillus gen. nov.
5.2. Description of Aliineobacillus hadale sp. nov.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jamieson, A.J.; Stewart, H.A. Hadal zones of the Northwest Pacific Ocean. Prog. Oceanogr. 2021, 190, 102477. [Google Scholar] [CrossRef]
- Gvirtzman, Z.; Stern, R.J. Bathymetry of mariana trench-arc system and formation of the challenger deep as a consequence of weak plate coupling. Tectonics 2004, 23, TC2011. [Google Scholar] [CrossRef]
- Liu, R.; Wang, L.; Wei, Y.; Fang, J. The hadal biosphere: Recent insights and new directions. Deep Sea Res. Part II Top. Stud. Oceanogr. 2018, 155, 11–18. [Google Scholar] [CrossRef]
- Tamburini, C.; Boutrif, M.; Garel, M.; Colwell, R.R.; Deming, J.W. Prokaryotic responses to hydrostatic pressure in the ocean—A review. Environ. Microbiol. 2013, 15, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, S.; Hirai, M.; Matsui, Y.; Makabe, A.; Minegishi, H.; Tsuda, M.; Juliarni; Rastelli, E.; Danovaro, R.; Corinaldesi, C.; et al. Microbial community and geochemical analyses of trans-trench sediments for understanding the roles of hadal environments. ISME J. 2020, 14, 740–756. [Google Scholar] [CrossRef]
- Shu, W.-S.; Huang, L.-N. Microbial diversity in extreme environments. Nat. Rev. Microbiol. 2022, 20, 219–235. [Google Scholar] [CrossRef]
- Flores, E.; Fernández-Urruzola, I.; Cantarero, S.I.; Pizarro-Koch, M.; Zabel, M.; Sepúlveda, J.; Ulloa, O. Particulate organic matter in the Atacama Trench: Tracing sources and possible transport mechanisms to the hadal seafloor. J. Geophys. Res. Biogeosci. 2023, 128, e2023JG007401. [Google Scholar] [CrossRef]
- Stief, P.; Schauberger, C.; Becker, K.W.; Elvert, M.; Balmonte, J.P.; Franco-Cisterna, B.; Middelboe, M.; Glud, R.N. Hydrostatic pressure induces transformations in the organic matter and microbial community composition of marine snow particles. Commun. Earth Environ. 2023, 4, 377. [Google Scholar] [CrossRef]
- Froelich, P.N.; Klinkhammer, G.P.; Bender, M.L.; Luedtke, N.A.; Heath, G.R.; Cullen, D.; Dauphin, P.; Hammond, D.; Hartman, B.; Maynard, V. Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: Suboxic diagenesis. Geochim. Cosmochim. Acta 1979, 43, 1075–1090. [Google Scholar] [CrossRef]
- Canfield, D.E.; Kristensen, E.; Thamdrup, B. Aquatic geomicrobiology. Adv. Mar. Biol. 2005, 48, 1–599. [Google Scholar]
- Lam, P.; Kuypers, M.M.M. Microbial Nitrogen Cycling Processes in Oxygen Minimum Zones. Annu. Rev. Mar. Sci. 2011, 3, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Callbeck, C.M.; Lavik, G.; Ferdelman, T.G.; Fuchs, B.; Gruber-Vodicka, H.R.; Hach, P.F.; Littmann, S.; Schoffelen, N.J.; Kalvelage, T.; Thomsen, S.; et al. Oxygen minimum zone cryptic sulfur cycling sustained by offshore transport of key sulfur oxidizing bacteria. Nat. Commun. 2018, 9, 1729. [Google Scholar] [CrossRef] [PubMed]
- Yayanos, A.A. Microbiology to 10,500 meter in the deep sea. Annu. Rev. Microbiol. 1995, 49, 777–805. [Google Scholar] [CrossRef] [PubMed]
- Glud, R.N.; Berg, P.; Thamdrup, B.; Larsen, M.; Stewart, H.A.; Jamieson, A.J.; Glud, A.; Oguri, K.; Sanei, H.; Rowden, A.A.; et al. Hadal trenches are dynamic hotspots for early diagenesis in the deep sea. Commun. Earth Environ. 2021, 2, 21. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Yutin, N.; Wolf, Y.I.; Vera Alvarez, R.; Koonin, E.V. Conservation and evolution of the sporulation gene set in diverse members of the firmicutes. J. Bacteriol. 2022, 204, e0007922. [Google Scholar] [CrossRef]
- Song, Y.C.; Holland, S.I.; Lee, M.; Chen, G.; Löffler, F.E.; Manefield, M.J.; Hugenholtz, P.; Kappler, U. A comparative genome analysis of the Bacillota (Firmicutes) class Dehalobacteriia. Microb. Genom. 2023, 9, 001039. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.K.; Poudel, S.; Yee, N.; Goff, J.L. Deeply branching Bacillota species exhibit atypical gram-negative staining. Microbiol. Spectr. 2024, 12, e0073224. [Google Scholar] [CrossRef]
- Jamieson, A. The Hadal Zone: Life in the Deepest Oceans; Cambridge University Press: Cambridge, UK, 2015; pp. 1–372. [Google Scholar]
- Nagel, M.; Andreesen, J.R. Bacillus niacini sp. nov., a Nicotinate-Metabolizing Mesophile Isolated from Soil. Int. J. Syst. Evol. Microbiol. 1991, 41, 134–139. [Google Scholar] [CrossRef]
- Heyrman, J.; Vanparys, B.; Logan, N.A.; Balcaen, A.; Rodríguez-Díaz, M.; Felske, A.; De Vos, P. Bacillus novalis sp. nov., Bacillus vireti sp. nov., Bacillus soli sp. nov., Bacillus bataviensis sp. nov. and Bacillus drentensis sp. nov., from the Drentse A grasslands. Int. J. Syst. Evol. Microbiol. 2004, 54, 47–57. [Google Scholar] [CrossRef]
- Liu, G.-H.; Narsing Rao, M.P.; Chen, Q.-Q.; Shi, H.; Che, J.-M.; Liu, B.; Li, W.-J. Neobacillus rhizophilus sp. nov. and Neobacillus citreus sp. nov., isolated from the citrus rhizosphere soil. Arch. Microbiol. 2022, 204, 281. [Google Scholar] [CrossRef]
- Gupta, R.S.; Patel, S.; Saini, N.; Chen, S. Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: Description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int. J. Syst. Evol. Microbiol. 2020, 70, 5753–5798. [Google Scholar]
- Yu, L.; Tang, X.; Wei, S.; Qiu, Y.; Xu, X.; Xu, G.; Wang, Q.; Yang, Q. Two novel species of the family Bacillaceae: Oceanobacillus piezotolerans sp. nov. and Bacillus piezotolerans sp. nov., from deep-sea sediment samples of Yap Trench. Int. J. Syst. Evol. Microbiol. 2019, 69, 3022–3030. [Google Scholar] [CrossRef]
- Minnikin, D.E.; O’Donnell, A.G.; Goodfellow, M.; Alderson, G.; Athalye, M.; Schaal, A.; Parlett, J.H. An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Methods 1984, 2, 233–241. [Google Scholar] [CrossRef]
- Tindall, B.J. Lipid composition of Halobacterium lacusprofundi. FEMS Microbiol. Lett. 1990, 66, 199–202. [Google Scholar] [CrossRef]
- Athalye, M.; Noble, W.C.; Mallet, A.I.; Minnikin, D.E. Gas chromatography-mass spectrometry of mycolic acids as a tool in the identification of medically important coryneform bacteria. J. Gen. Microbiol. 1984, 130, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Kato, C.; Runko, G.M.; Nogi, Y.; Hori, T.; Li, J.; Morono, Y.; Inagaki, F. Predominance of viable spore-forming piezophilic bacteria in high-pressure enrichment cultures from ~1.5 to 2.4 km-deep coal-bearing sediments below the ocean floor. Front. Microbiol. 2017, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Vera Alvarez, R.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef] [PubMed]
- Elbourne, L.D.; Tetu, S.G.; Hassan, K.A.; Paulsen, I.T. TransportDB 2.0: A database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 2017, 45, D320–D324. [Google Scholar] [CrossRef]
- Ye, J.; McGinnis, S.; Madden, T.L. BLAST:Improvements for better sequence analysis. Nucleic Acids Res. 2006, 34, W6–W9. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32, 929–931. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Klenk, H.-P.; Göker, M. Taxonomic use of DNA G+C content and DNA–DNA hybridization in the genomic age. Int. J. Syst. Evol. Microbiol. 2014, 64, 352–356. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Csűös, M. Count: Evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 2010, 26, 1910–1912. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Zhang, Q.; Narsing Rao, M.P.; Liu, G.-H.; Che, J.-M.; Lei, M.; Liu, B.; Li, W.-J.; Zhou, S.-G. Neobacillus sedimentimangrovi sp. nov., a Thermophilic Bacterium Isolated from Mangrove Sediment. Curr. Microbiol. 2021, 78, 1039–1044. [Google Scholar] [CrossRef]
- Tang, X.; Yu, L.; Yi, Y.; Wang, J.; Wang, S.; Meng, C.; Liu, S.; Hao, Y.; Zhang, Y.; Cao, X.; et al. Phylogenomic analysis reveals a two-stage process of the evolutionary transition of Shewanella from the upper ocean to the hadal zone. Environ. Microbiol. 2021, 23, 744–756. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Ranges (optimum) for growth: | ||||||
| Temperature(°C) | 15–45 | 10–45 | 16–50 | 10–50 | 0–50 | 16–50 |
| NaCl (w/v, %) | 0–3 | 0–10 | 0–5 | 0–15 | 0–15 | 0–5 |
| pH | 5–8 | 6.0–9.5 | 6.0–10.0 | 6.0–12.0 | 6.0–12.0 | 6.0–10.0 |
| Fatty acids: | ||||||
| anteiso-C15:0 | 5.5 | 33.5 | 21.8 | 12.8 | 12.5 | 20.5 |
| iso-C17:1ω5ϲ | 5.5 | − | − | − | − | − |
| iso-C17:0 | 6.4 | 2.6 | 2.6 | 1.5 | 2.7 | 1.4 |
| C16:1ω7c alcohol | 0.3 | 2.4 | 3.1 | 5.0 | 3.4 | 2.3 |
| anteiso-C16:0 | 0.2 | − | − | − | − | − |
| anteiso-C17:1 A | 1.9 | − | − | − | − | − |
| iso-C13:0 | 4.3 | 0.2 | − | − | − | − |
| Acid production from: | ||||||
| glycerol | w | − | w | ND | ND | − |
| D-cellobiose | w | − | + | ND | ND | − |
| D-turanose | + | − | + | ND | ND | v |
| D-mannitol | + | − | + | ND | ND | − |
| L-fucose | w | v | − | ND | ND | − |
| DNA G + C content (mol%): | 40.90 | 40.10 | 40.10 | 40.80 | 41.10 | 39.40 |
| Items | Description |
|---|---|
| Size (bp) | 3,735,591 |
| G + C content (%) | 40.9 |
| Genes assigned to COG | 3075 |
| Protein-coding genes | 3614 |
| 16S rRNA | 9 |
| 23S rRNA | 9 |
| 5S rRNA | 9 |
| rRNA operons | 9 |
| tRNA genes | 86 |
| Gene islands | 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Liu, J.; Liu, Y.; Wu, W.; Wang, J.; Wei, Y. Comparative Genomics Reveals Evidence of the Genome Reduction and Metabolic Potentials of Aliineobacillus hadale Isolated from Challenger Deep Sediment of the Mariana Trench. Microorganisms 2025, 13, 132. https://doi.org/10.3390/microorganisms13010132
Yang S, Liu J, Liu Y, Wu W, Wang J, Wei Y. Comparative Genomics Reveals Evidence of the Genome Reduction and Metabolic Potentials of Aliineobacillus hadale Isolated from Challenger Deep Sediment of the Mariana Trench. Microorganisms. 2025; 13(1):132. https://doi.org/10.3390/microorganisms13010132
Chicago/Turabian StyleYang, Shaofeng, Jie Liu, Yang Liu, Weichao Wu, Jiahua Wang, and Yuli Wei. 2025. "Comparative Genomics Reveals Evidence of the Genome Reduction and Metabolic Potentials of Aliineobacillus hadale Isolated from Challenger Deep Sediment of the Mariana Trench" Microorganisms 13, no. 1: 132. https://doi.org/10.3390/microorganisms13010132
APA StyleYang, S., Liu, J., Liu, Y., Wu, W., Wang, J., & Wei, Y. (2025). Comparative Genomics Reveals Evidence of the Genome Reduction and Metabolic Potentials of Aliineobacillus hadale Isolated from Challenger Deep Sediment of the Mariana Trench. Microorganisms, 13(1), 132. https://doi.org/10.3390/microorganisms13010132