

Characterization by MALDI-TOF MS and 16S rRNA Gene Sequencing of Aerobic Endospore-Forming Bacteria Isolated from Pharmaceutical Facility in Rio de Janeiro, Brazil

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Identification by MALDI-TOF MS
2.3. 16S rRNA Gene Sequencing and Analysis
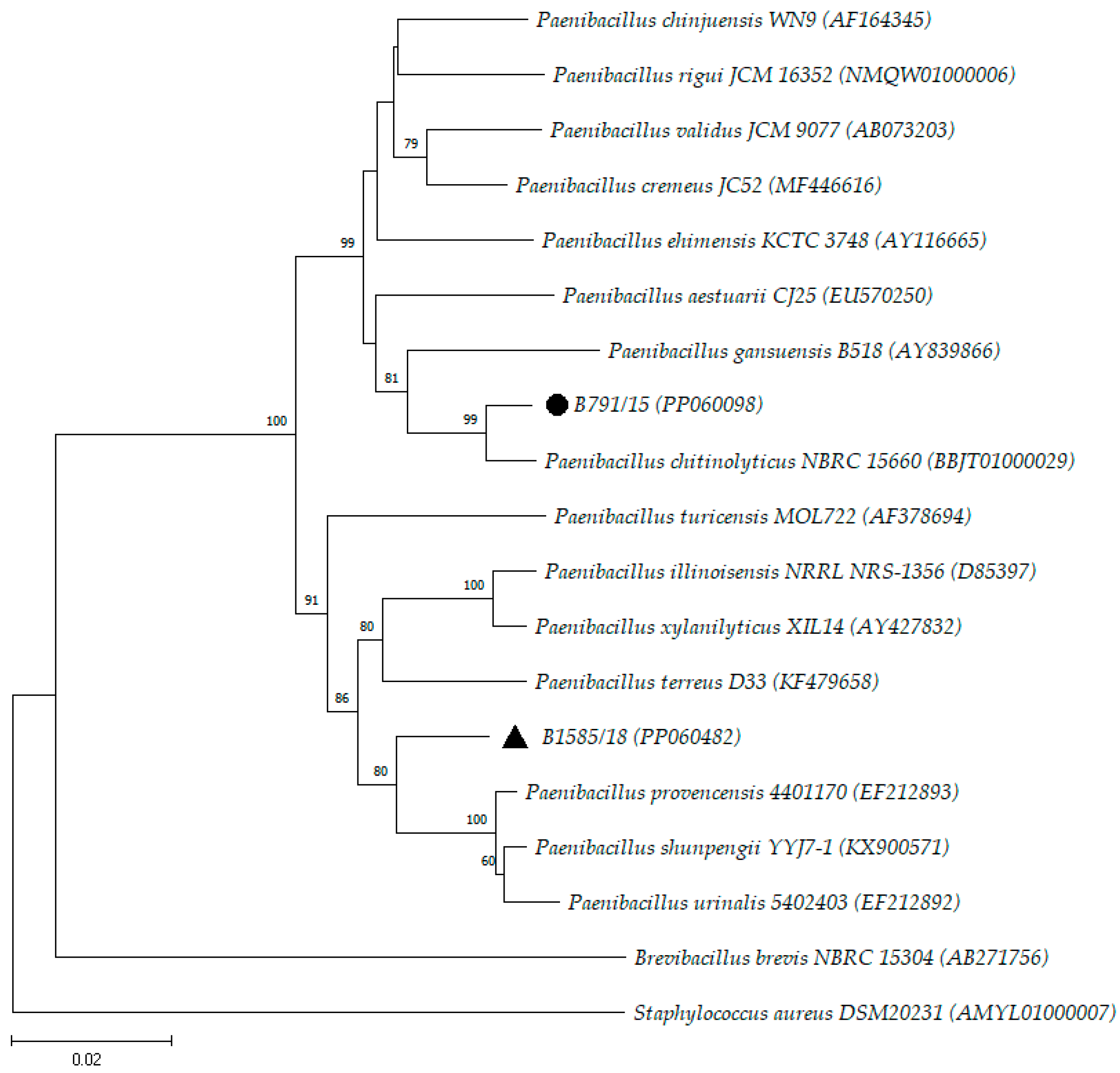
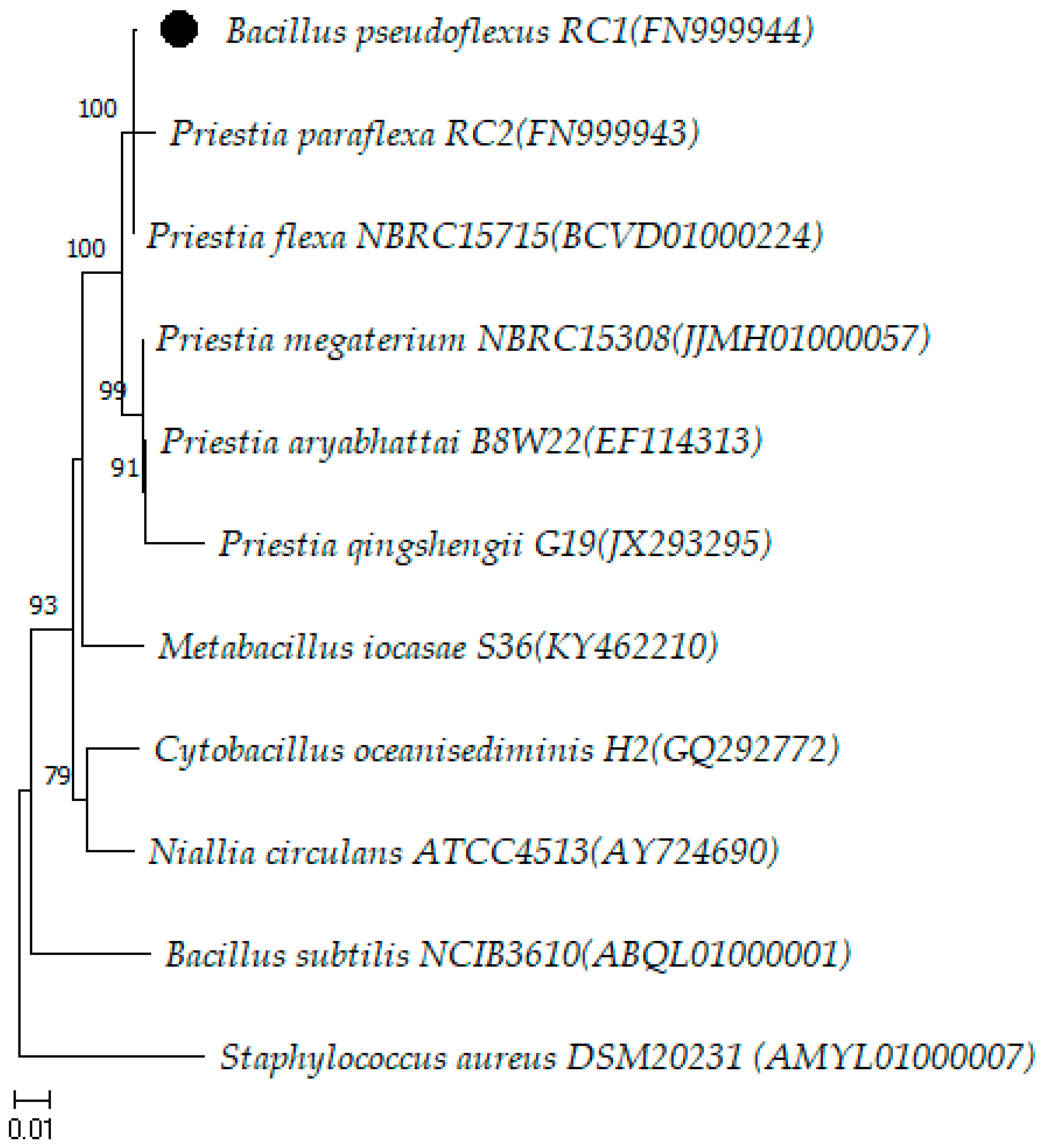
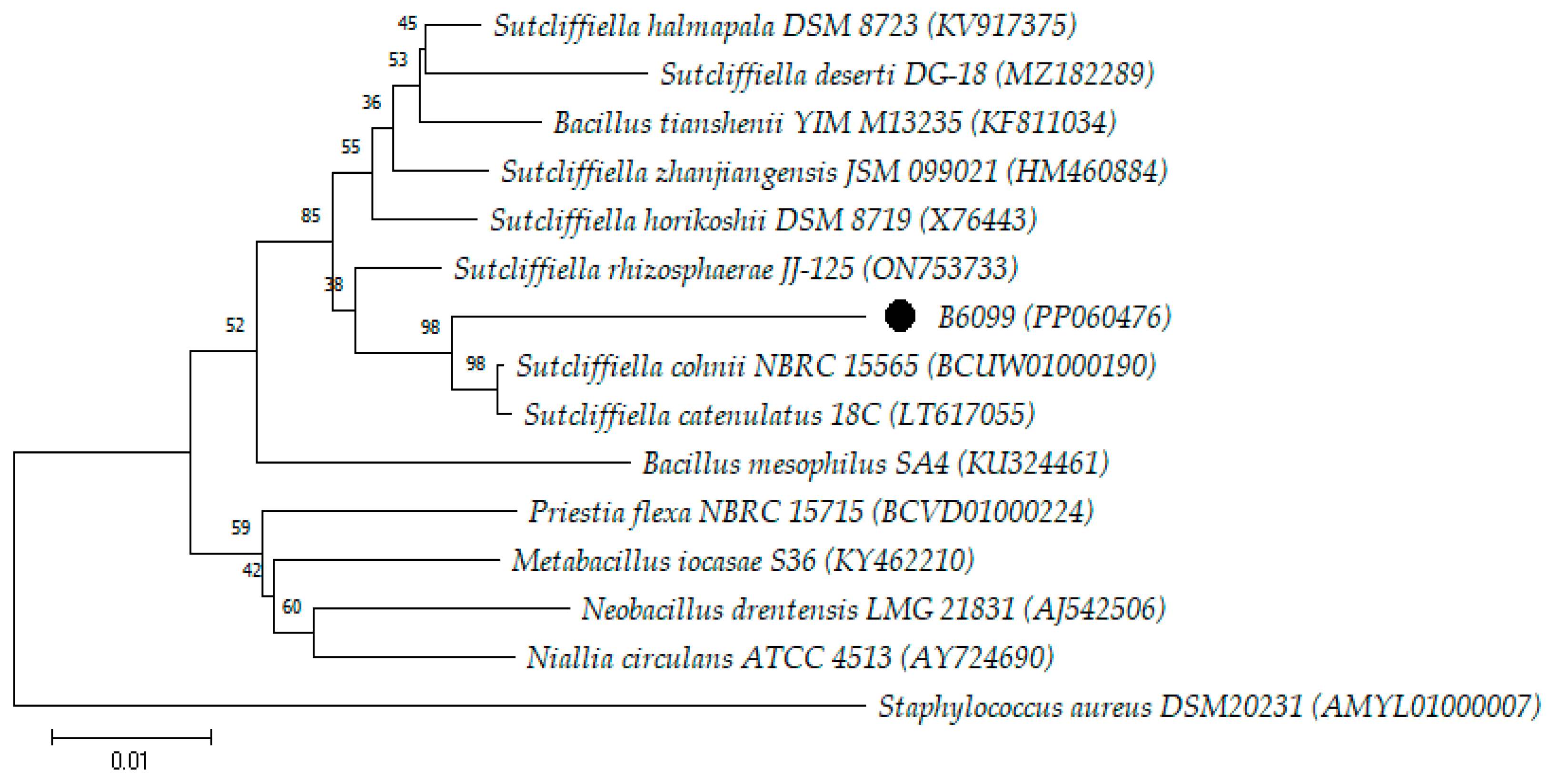
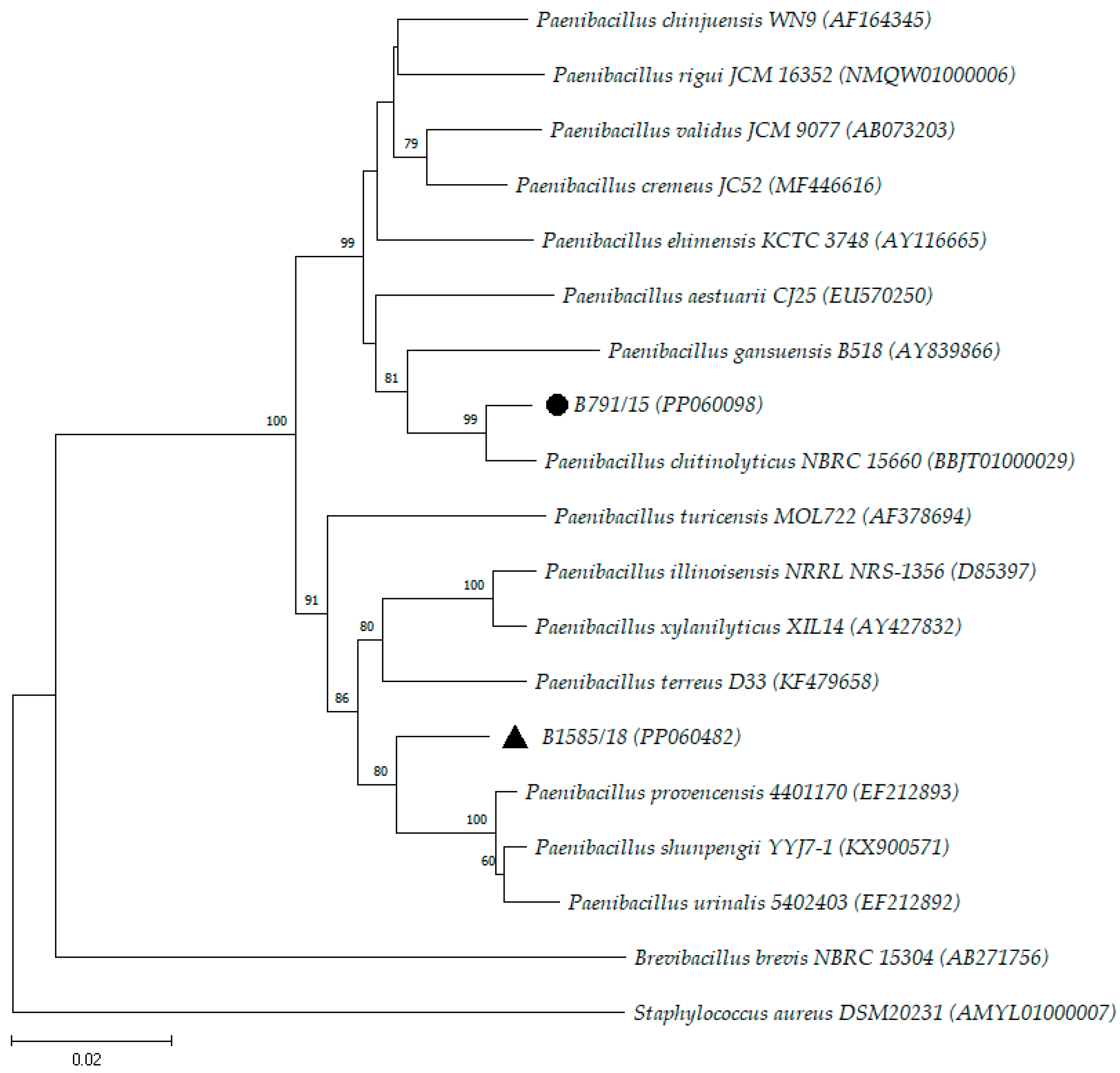
3. Results and Discussion
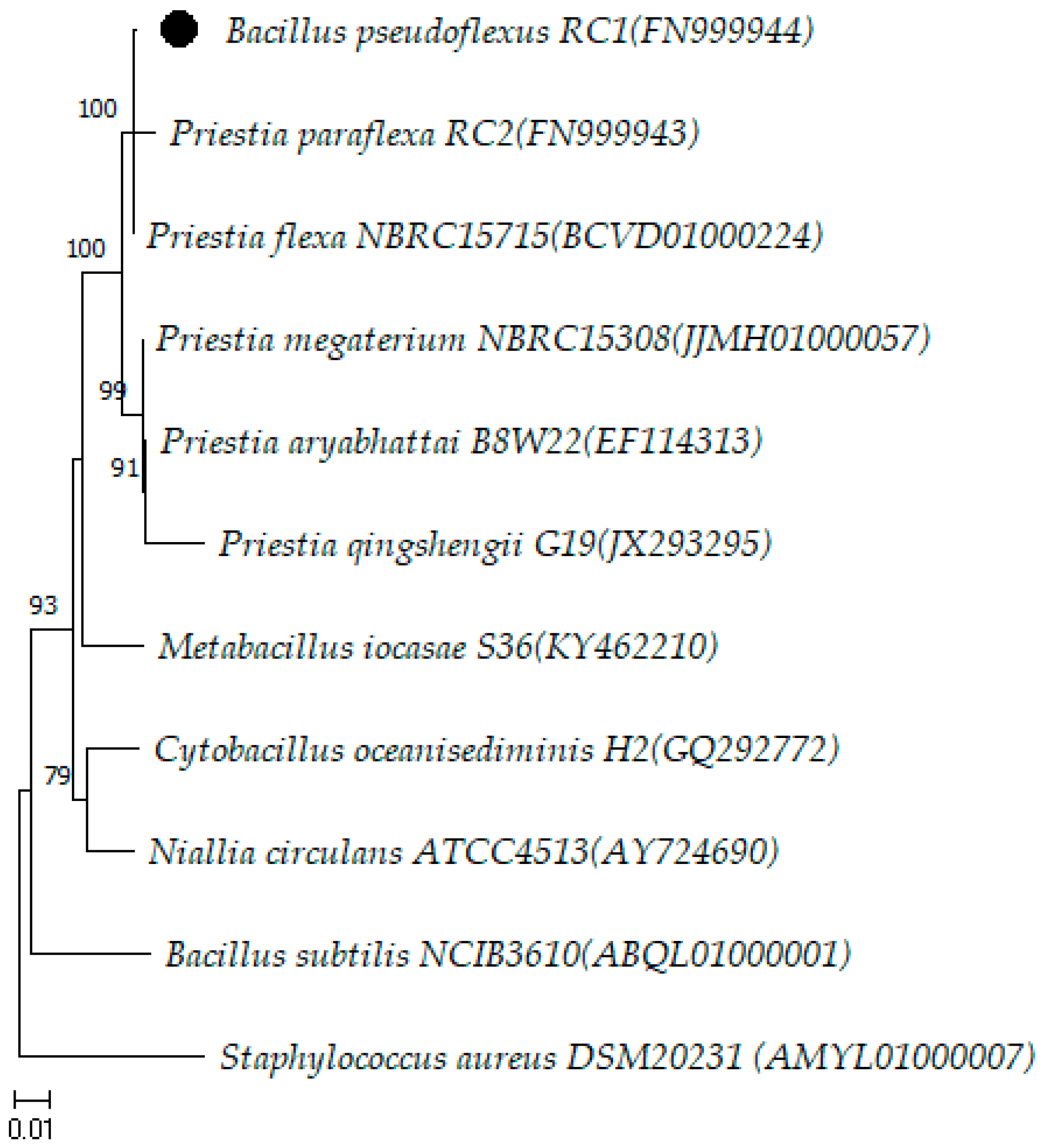
Description of Priestia pseudoflexus sp. nov.
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Röder, F.; Sandle, T. Microbial Contamination in Water Systems. PDA J. Pharm. Sci. Technol. 2022, 76, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Sandle, T. A review of cleanroom microflora: Types, trends, and patterns. PDA J. Pharm. Sci. Technol. 2011, 65, 392–403. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. European Medicines Agency. European Union guidelines for good manufacturing practice for medicinal products for human and veterinary use. In The Rules Governing Medicinal Products in the European Union; Annex 1: Manufacture of Sterile Medicinal Products; European Medicines Agency: Brussels, Belgium, 2022; Volume 4. [Google Scholar]
- National Health Surveillance Agency (ANVISA). Resolution of the collegiate board—RDC no 609, of 9 March 2022. In Brazilian Pharmacopoeia, 6th ed.; ANVISA: Brasilia, Brazil, 2019; Volume 1. Available online: https://www.gov.br/anvisa/pt-br/assuntos/pharmacopoeia/pharmacopoeia-brazilian (accessed on 10 January 2024).
- United States Pharmacopeial Convention (USP). The United States Pharmacopeia, 43rd ed.; United States Pharmacopeial Convention Inc.: Rockville, MD, USA, 2021; Available online: http://www.uspnf.com/uspnf/login (accessed on 24 August 2022).
- Costa, L.V.D.; Miranda, R.V.D.S.L.; Fonseca, E.L.; Gonçalves, N.P.; Reis, C.M.F.; Frazão, A.M.; Cruz, F.V.; Brandão, M.L.L.; Ramos, J.N.; Vieira, V.V. Assessment of VITEK® 2, MALDI-TOF MS and full gene 16S rRNA sequencing for aerobic endospore-forming bacteria isolated from a pharmaceutical facility. J. Microbiol. Methods 2022, 194, 106419. [Google Scholar] [CrossRef] [PubMed]
- Miranda, R.V.D.S.L.; da Costa, L.V.; Albuquerque, L.S.; Dos Reis, C.M.F.; Braga, L.M.P.D.S.; de Andrade, J.M.; Ramos, J.N.; Mattoso, J.M.V.; Forsythe, S.J.; Brandão, M.L.L. Identification of Sutcliffiella horikoshii strains in an immunobiological pharmaceutical industry facility. Lett. Appl. Microbiol. 2023, 76, ovad056. [Google Scholar] [CrossRef]
- Celandroni, F.; Salvetti, S.; Gueye, S.A.; Mazzantini, D.; Lupetti, A.; Senesi, S.; Ghelardi, E. Identification and Pathogenic Potential of Clinical Bacillus and Paenibacillus Isolates. PLoS ONE 2016, 11, e0152831. [Google Scholar] [CrossRef] [PubMed]
- Sakhno, N.G.; Gunar, O.V. Microbial Identification Methods in Pharmaceutical Analysis: Comparison and Evaluation. Mathews J. Pharm. Sci. 2016, 1, 1–8. [Google Scholar]
- Kopcakova, A.; Stramova, Z.; Kvasnova, S.; Godany, A.; Perhacova, Z.; Pristas, P. Need for database extension for reliable identification of bacteria from extreme environments using MALDI TOF mass spectrometry. Chem. Pap. 2014, 68, 1435–1442. [Google Scholar] [CrossRef]
- Costa, L.V.D.; Miranda, R.V.D.S.L.; Reis, C.M.F.; Andrade, J.M.; Cruz, F.V.; Frazão, A.M.; Fonseca, E.L.; Ramos, J.N.; Brandão, M.L.L.; Vieira, V.V. MALDI-TOF MS database expansion for identification of Bacillus and related genera isolated from a pharmaceutical facility. J. Microbiol. Methods 2022, 203, 106625. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11.0.13. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Gupta, R.S.; Patel, S.; Saini, N.; Chen, S. Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: Description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int. J. Syst. Evol. Microbiol. 2020, 70, 5753–5798. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Gupta, R.S. A phylogenomic and comparative genomic framework for resolving the polyphyly of the genus Bacillus: Proposal for six new genera of Bacillus species, Peribacillus gen. nov., Cytobacillus gen. nov., Mesobacillus gen. nov., Neobacillus gen. nov., Metabacillus gen. nov. and Alkalihalobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 406–438. [Google Scholar] [CrossRef] [PubMed]
- TeKippe, E.M.; Burnham, C.-A.D. Evaluation of the Bruker Biotyper and VITEK MS MALDI-TOF MS systems for the identification of unusual and/or difficult-to-identify microorganisms isolated from clinical specimens. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Mezian, L.; Chincha, A.I.A.; Vecchione, A.; Ghelardi, E.; Bonatto, J.M.C.; Marsaioli, A.J.; Campelo, P.H.; Benamar, I.; Moussaoui, A.A.; Sant’Ana, A.S.; et al. Aerobic spore-forming bacteria in powdered infant formula: Enumeration, identification by MALDI-TOF mass spectrometry (MS), presence of toxin genes and rpoB gene typing. Int. J. Food Microbiol. 2022, 368, 109613. [Google Scholar] [CrossRef]
- Orem, J.C.; Silva, W.M.C.; Raiol, T.; Magalhães, M.I.; Martins, P.H.; Cavalcante, D.A.; Kruger, R.H.; Brigido, M.M.; De-Souza, M.T. Phylogenetic diversity of aerobic spore-forming Bacillalles isolated from Brazilian soils. Int. Microbiol. 2019, 22, 511–520. [Google Scholar] [CrossRef]
- Pereira, P.P.; Torres, T.G.A.; Fernandez, M.; Blanch, A.R.; Gonzalez, P.S.; Agostini, E. Polyphasic characterization and identification of the bioremediation agent Bacillus sp. SFC 500-1E. Genomics 2020, 112, 4525–4535. [Google Scholar] [CrossRef] [PubMed]
- Chandna, P.; Mayilraj, S.; Kuhad, R.C. Bacillus pseudoflexus sp. Nov., a moderately halophilic bacterium isolated from compost. Ann. Microbiol. 2016, 66, 895–905. [Google Scholar] [CrossRef]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- Priest, F.G.; Goodfellow, M.; Todd, C. A Numerical Classification of the Genus Bacillus. J. Gen. Microbiol. 1988, 134, 1847–1882. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Strain | Origin | VITEK® MS (%) | MALDI Biotyper® (Score) |
|---|---|---|---|
| B6081 | Air monitoring | Paenibacillus spp. (83.80) | P. glucanolyticus (1.88) |
| B6099 | Water vapor | NI | B. cohnii (1.94) |
| B6114 | Water vapor | NI | NI |
| B6389 | Operator monitoring | NI | NI |
| B6444 | Bioburden test | Ureibacillus spp. (84.60) | NI |
| B791/15 | Potable water | NI | NI |
| B1513/15 | Sterility test | NI | NI |
| B636/16 | Sterility test | NI | NI |
| B669/18 | Operator monitoring | NI | NI |
| B1439/18 | Bioburden test | NI | B. endophyticus (1.74) |
| B1567/18 | Potable water | NI | NI |
| B1585/18 | Air monitoring | NI | NI |
| B554/19 | Bioburden test | NI | NI |
| B773/19 | Bioburden test | NI | NI |
| B1124/19 | Bioburden test | NI | NI |
| B1665/18 | Bioburden test | P. validus (99.90) | P. validus (2.21) |
| B1710/18 | Bioburden test | B. licheniformis (99.90) | B. licheniformis (2.14) |
| B1711/18 | Bioburden test | Bacillus spp. (81.20) | B. siralis (2.15) |
| B1819/18 | Bioburden test | B. cereus group (99.90) | B. cereus (2.09) |
| B1847/18 | Bioburden test | B. cereus group (99.90) | B. cereus (2.15) |
| B1972/18 | Bioburden test | B. cereus group (89.90) | B. cereus (2.14) |
| B1192/19 | Purified water | L. fusiformis/sphaericus (99.00) | L. fusiformis (2.06) |
| B1209/19 | Bioburden test | B. flexus (84.60) | B. flexus (2.26) |
| B1213/19 | Sterility test | B. flexus (75.60) | B. flexus (2.23) |
| B097/20 | Bioburden test | B. flexus (88.20) | B. flexus (2.36) |
| Strain | NCBI ¹ Access Number | bp ² | Species (% of Similarity) |
|---|---|---|---|
| B6081 | PP059127 | 1501 | P. lautus (99.25) |
| B791/15 | PP060098 | 1401 | P. chitinolyticus (98.50) |
| B1585/18 | PP060482 | 1488 | P. provencensis (97.04) |
| B6099 | PP060476 | 1447 | S. cohnii (96.37) |
| B6389 | PP059202 | 1426 | M. idriensis (99.35) |
| B554/19 | PP060516 | 1470 | S. inulinus (98.83)/S. terrae (98.77) |
| B636/16 | PP060583 | 1432 | N. drentensis (99.42)/N. soli (99.16)/N. bataviensis (99.02)/N. vireti (99.01)/N. cucumis (98.95)/N. novalis (98.81) |
| B1439/18 | PP060597 | 1495 | P. filamentosa (99.86)/P. endophytica (99.32) |
| B773/19 | PP060628 | 1457 | P. flexa (99.86)/B. pseudoflexus (99.65)/P. paraflexa (98.84)/P. megaterium (98.83)/P. aryabhattai (98.69) |
| B6444 | PP060737 | 1462 | U. chungkukjangi (99.86)/U. sinduriensis (99.00) |
| B6114 | PP060738 | 1445 | B. paralicheniformis (99.79)/B. glycinifermentans (99.58)/B. haynesii (99.51)/B. sonorensis (99.24)/B. licheniformis (99.17)/B. aerius (98.75) |
| B1513/15 | PP060747 | 1443 | B. siamensis (99.93)/B. velezensis (99.93)/B. subtilis (99.79)/B. amyloliquefaciens (99.72)/B. nakamurai (99.65)/B. tequilensis (99.58)/B. cabrialesii (99.58)/B. inaquosorum (99.58)/B. stercoris (99.58)/B. vallismortis (99.51)/B. atrophaeus (99.44)/B. halotolerans (99.44)/B. spizizenii (99.44)/B. mojavensis (99.37) |
| B669/18 | PP060782 | 1492 | B.velezensis (99.93)/B. siamensis (99.86)/B. subtilis (99.59)/B. amyloliquefaciens (99.59)/B. nakamurai (99.59)/B. atrophaeus (99.39)/B. tequilensis (99.39)/B. halotolerans (99.39)/B. cabrialesii (99.39)/B. inaquosorum (99.39)/B. stercoris (99.39)/B. mojavensis (99.32)/B. vallismortis (99.32)/B. spizizenii (99.25) |
| B1567/18 | PP060923 | 1465 | B. paralicheniformis (99.66)/B. glycinifermentans (99.45)/B. haynesii (99.38)/B. licheniformis (99.11)/B. sonorensis (99.11)/B. aerius (98.70) |
| B1124/19 | PP060925 | 1480 | B. licheniformis (99.52)/B.haynesii (99.45)/B. sonorensis (99.32)/B. paralicheniformis (99.18)/B. aerius (99.11)/B. glycinifermentans (98.98)/B. swezeyi (98.77) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caldeira, N.G.S.; de Souza, M.L.S.; de Miranda, R.V.d.S.L.; da Costa, L.V.; Forsythe, S.J.; Zahner, V.; Brandão, M.L.L. Characterization by MALDI-TOF MS and 16S rRNA Gene Sequencing of Aerobic Endospore-Forming Bacteria Isolated from Pharmaceutical Facility in Rio de Janeiro, Brazil. Microorganisms 2024, 12, 724. https://doi.org/10.3390/microorganisms12040724
Caldeira NGS, de Souza MLS, de Miranda RVdSL, da Costa LV, Forsythe SJ, Zahner V, Brandão MLL. Characterization by MALDI-TOF MS and 16S rRNA Gene Sequencing of Aerobic Endospore-Forming Bacteria Isolated from Pharmaceutical Facility in Rio de Janeiro, Brazil. Microorganisms. 2024; 12(4):724. https://doi.org/10.3390/microorganisms12040724
Chicago/Turabian StyleCaldeira, Nathalia Gonçalves Santos, Maria Luiza Soares de Souza, Rebeca Vitória da Silva Lage de Miranda, Luciana Veloso da Costa, Stephen James Forsythe, Viviane Zahner, and Marcelo Luiz Lima Brandão. 2024. "Characterization by MALDI-TOF MS and 16S rRNA Gene Sequencing of Aerobic Endospore-Forming Bacteria Isolated from Pharmaceutical Facility in Rio de Janeiro, Brazil" Microorganisms 12, no. 4: 724. https://doi.org/10.3390/microorganisms12040724
APA StyleCaldeira, N. G. S., de Souza, M. L. S., de Miranda, R. V. d. S. L., da Costa, L. V., Forsythe, S. J., Zahner, V., & Brandão, M. L. L. (2024). Characterization by MALDI-TOF MS and 16S rRNA Gene Sequencing of Aerobic Endospore-Forming Bacteria Isolated from Pharmaceutical Facility in Rio de Janeiro, Brazil. Microorganisms, 12(4), 724. https://doi.org/10.3390/microorganisms12040724