
Genetic and Evolutionary Analysis of Porcine Deltacoronavirus in Guangxi Province, Southern China, from 2020 to 2023
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection and Detection of Specimens for PDCoV
2.2. Amplification and Sequencing
2.3. Sequence Comparison and Phylogenetic Analysis
2.4. Bayesian Temporal Dynamics Analysis
2.5. Genome Recombination Events Analysis
3. Results
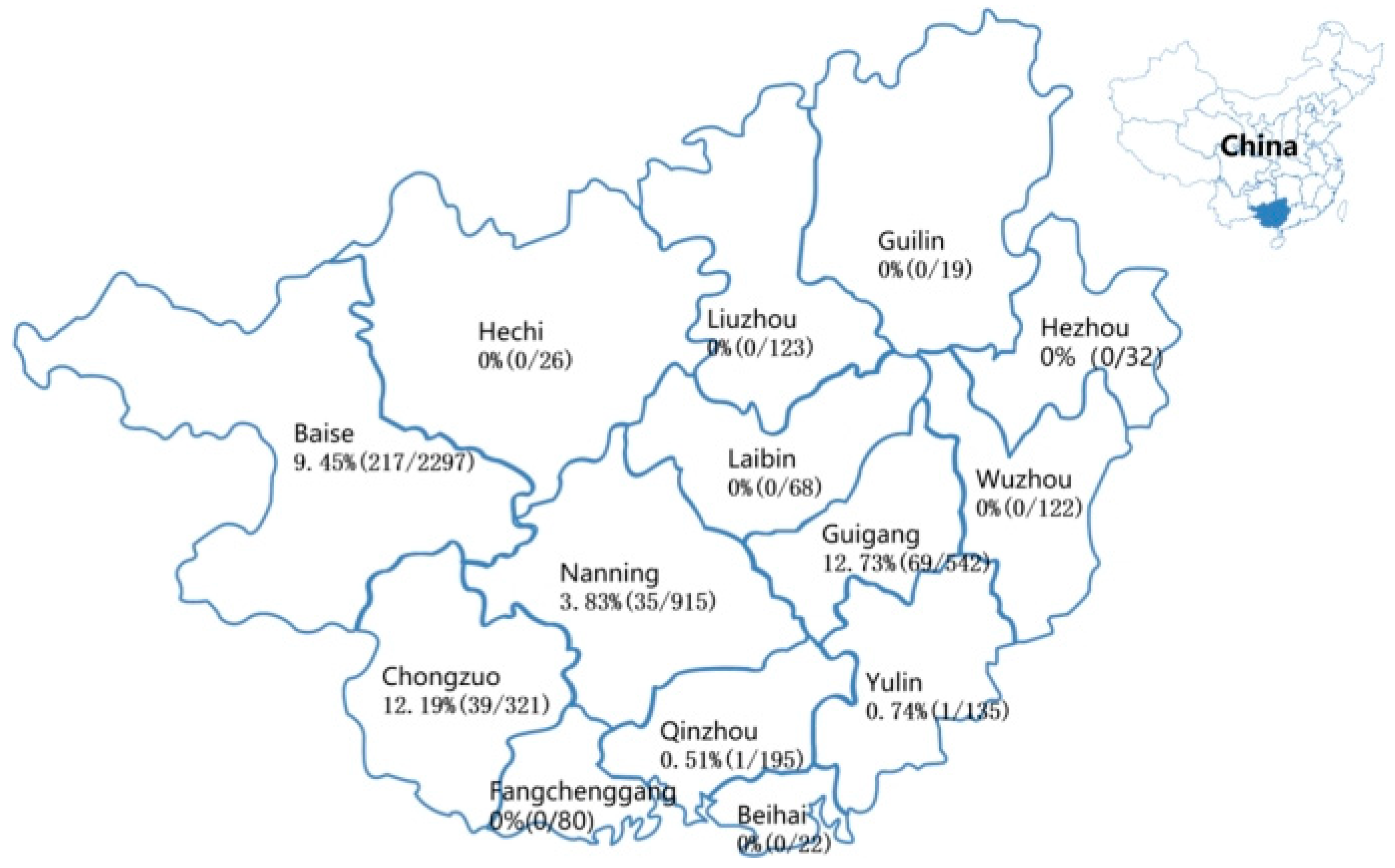
3.1. Detection Results of the Clinical Specimens
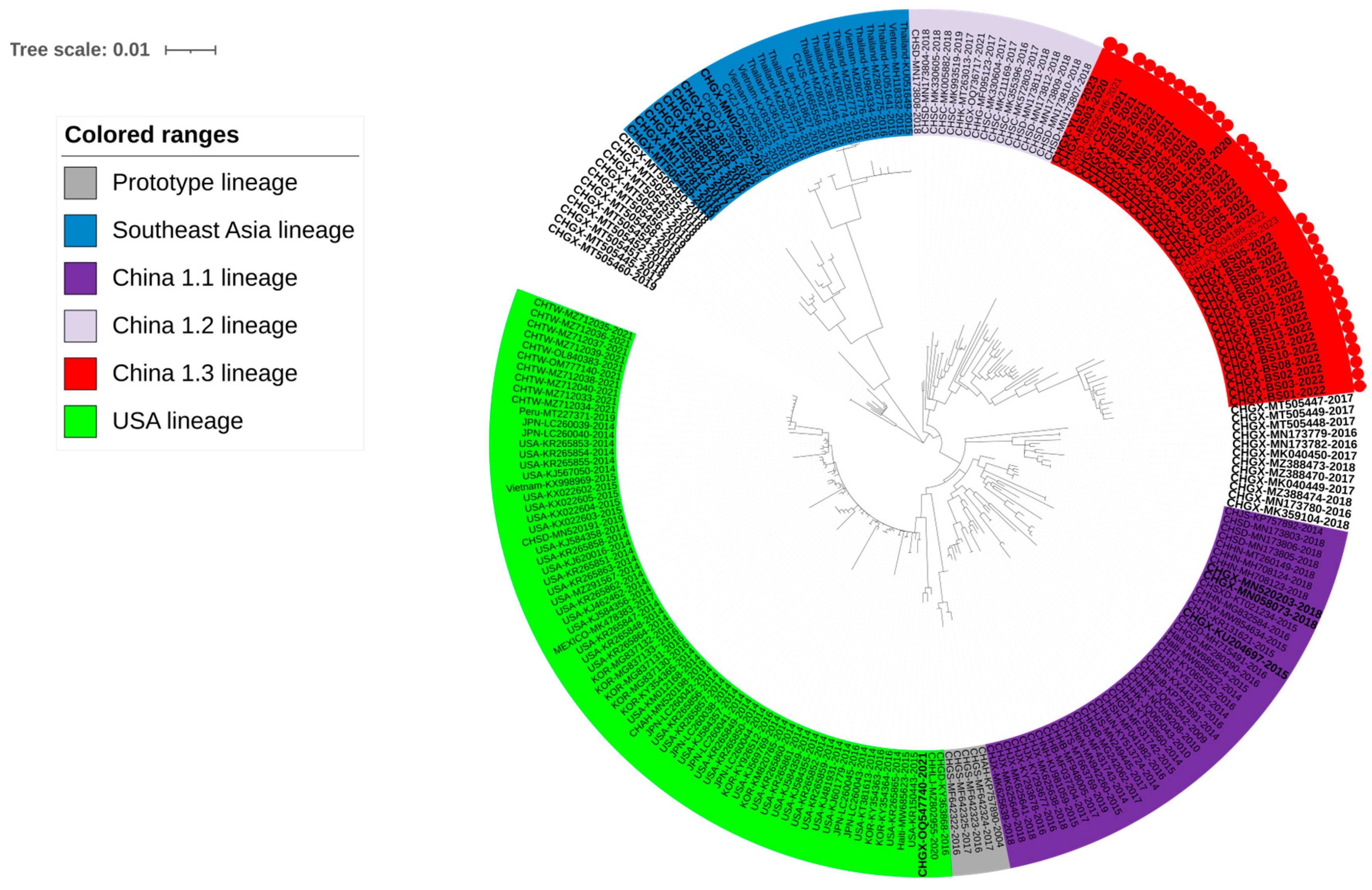
3.2. Phylogenetic Analysis Basing on S Gene Sequences
3.3. Phylogenetic Analysis Basing on M Gene Sequences
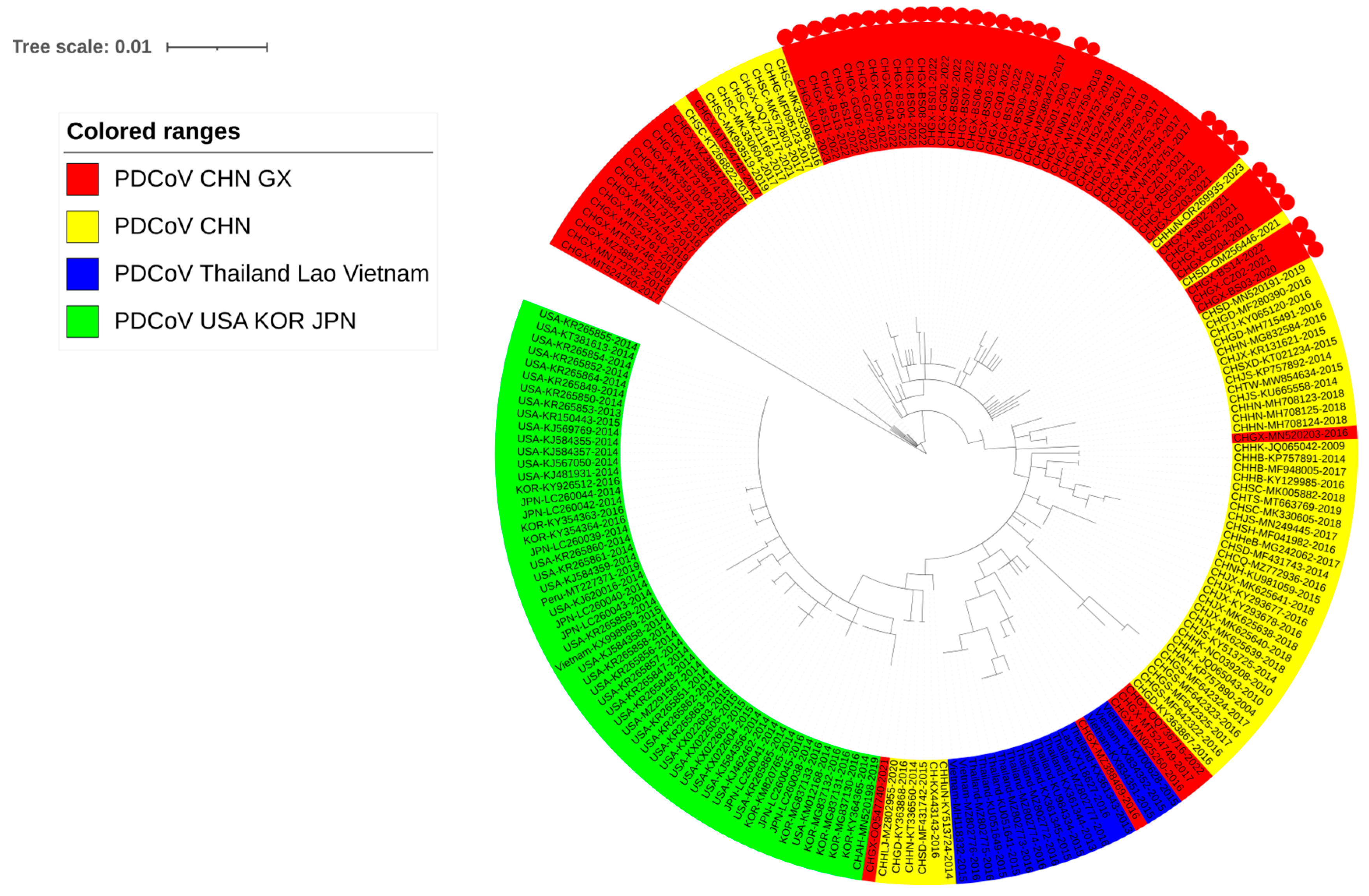
3.4. Phylogenetic Analysis Basing on N Gene Sequences
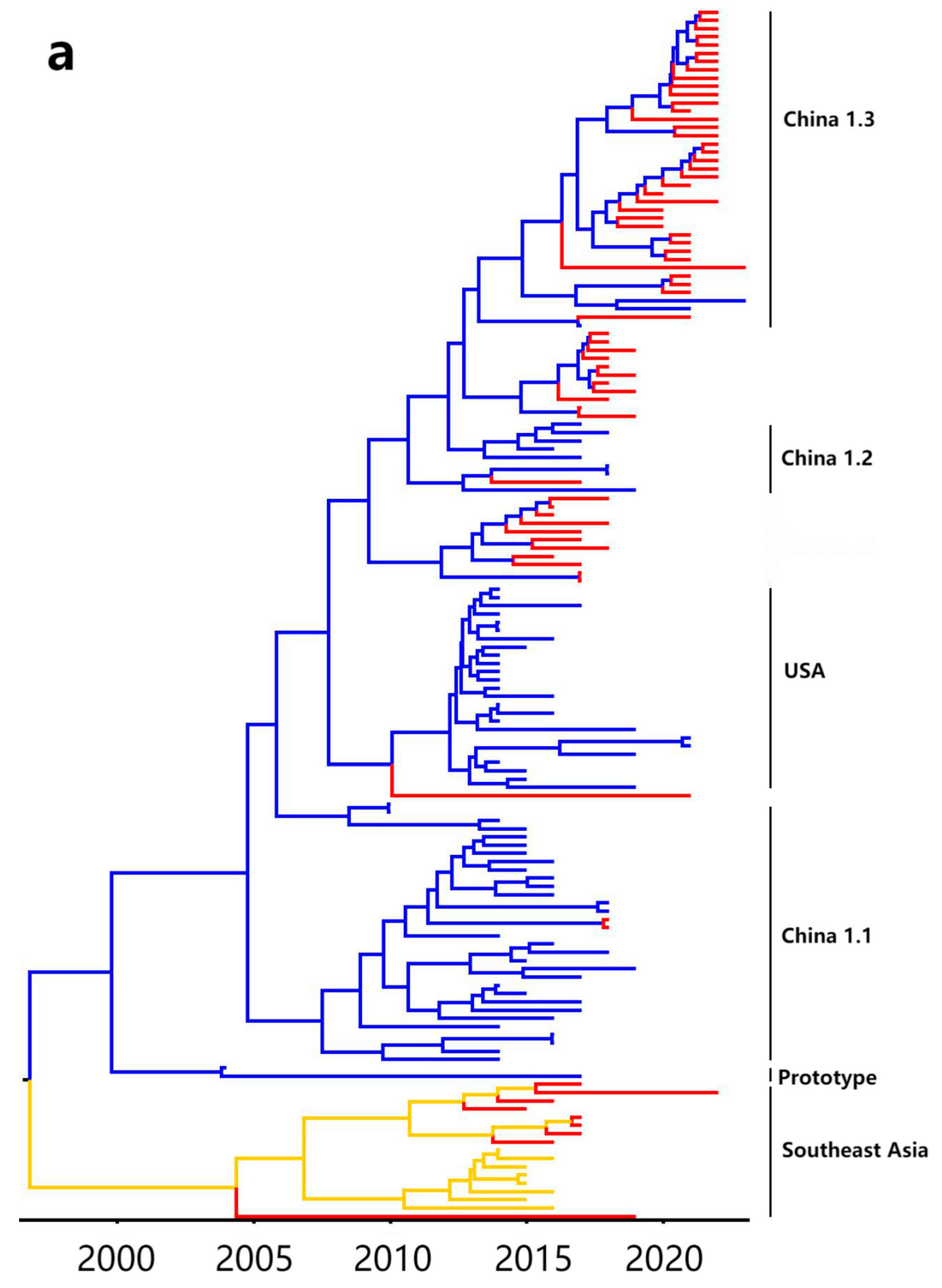
3.5. Bayesian Temporal Dynamics Analysis
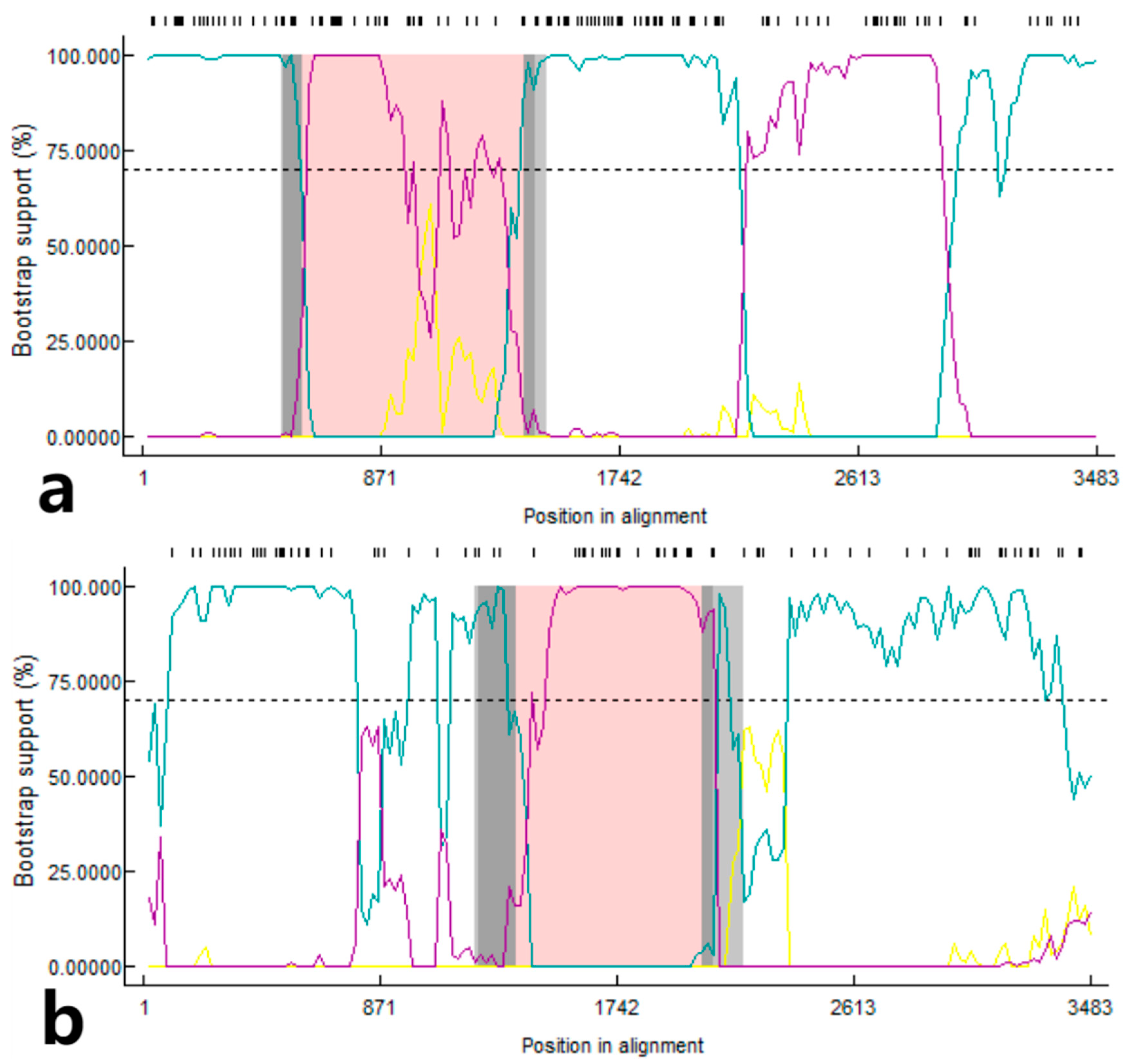
3.6. Recombination Analysis of S Gene
3.7. Genetic Evolution Rates of S, M, and N Genes
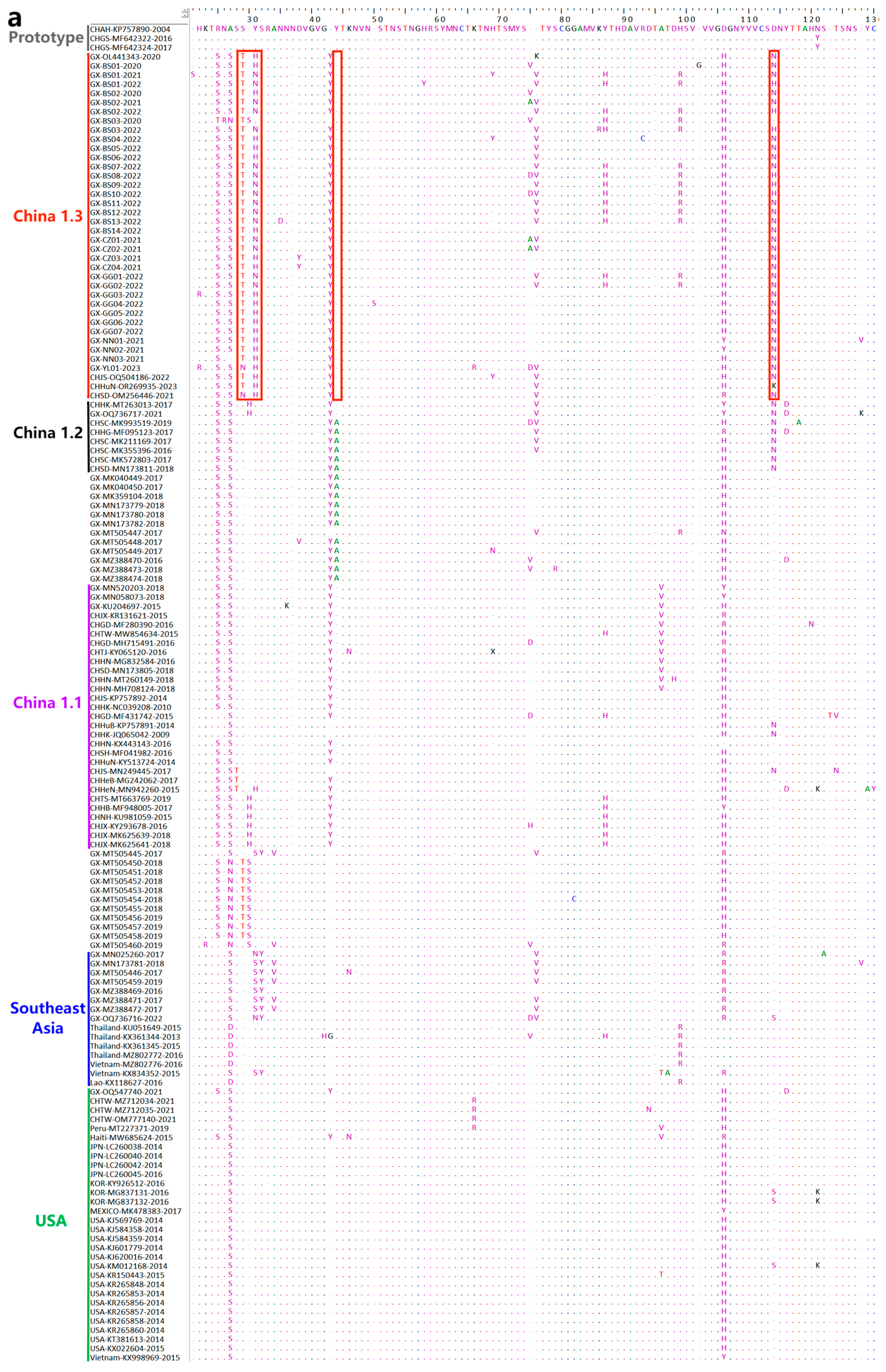
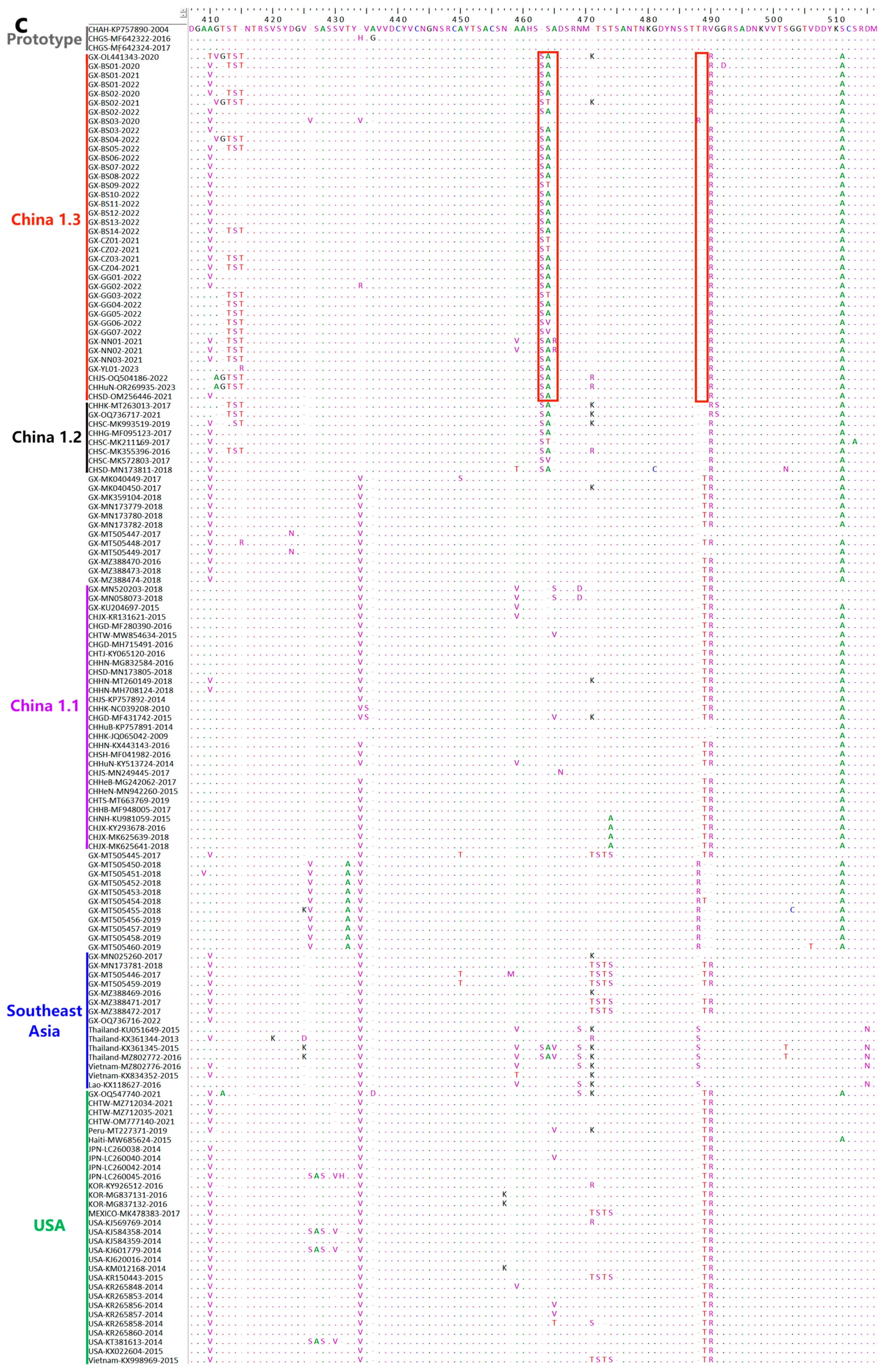
3.8. Deduence Analysis of the S Protein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Duan, C. An updated review of porcine deltacoronavirus in terms of prevalence, pathogenicity, pathogenesis and antiviral strategy. Front. Vet. Sci. 2022, 8, 811187. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Gauger, P.; Stafne, M.; Thomas, J.; Arruda, P.; Burrough, E.; Madson, D.; Brodie, J.; Magstadt, D.; Derscheid, R.; et al. Pathogenicity and pathogenesis of a United States porcine deltacoronavirus cell culture isolate in 5-day-old neonatal piglets. Virology 2015, 482, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Porcine deltacoronavirus: Overview of infection dynamics, diagnostic methods, prevalence and genetic evolution. Virus Res. 2016, 226, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Hu, H.; Saif, L.J. Porcine deltacoronavirus infection: Etiology, cell culture for virus isolation and propagation, molecular epidemiology and pathogenesis. Virus Res. 2016, 226, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; de Groot, R.J.; Haagmans, B.; Lau, S.K.P.; Neuman, B.W.; Perlman, S.; Sola, I.; van der Hoek, L.; Wong, A.C.P.; Yeh, S.H. ICTV virus taxonomy profile: Coronaviridae 2023. J. Gen. Virol. 2023, 104, 001843. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar]
- Lorsirigool, A.; Saeng-Chuto, K.; Madapong, A.; Temeeyasen, G.; Tripipat, T.; Kaewprommal, P.; Tantituvanont, A.; Piriyapongsa, J.; Nilubol, D. The genetic diversity and complete genome analysis of two novel porcine deltacoronavirus isolates in Thailand in 2015. Virus Genes 2017, 53, 240–248. [Google Scholar] [CrossRef]
- Le, V.P.; Song, S.; An, B.H.; Park, G.N.; Pham, N.T.; Le, D.Q.; Nguyen, V.T.; Vu, T.T.H.; Kim, K.S.; Choe, S.; et al. A novel strain of porcine deltacoronavirus in Vietnam. Arch. Virol. 2018, 163, 203–207. [Google Scholar] [CrossRef]
- Suzuki, T.; Shibahara, T.; Imai, N.; Yamamoto, T.; Ohashi, S. Genetic characterization and pathogenicity of Japanese porcine deltacoronavirus. Infect. Genet. Evol. 2018, 61, 176–182. [Google Scholar] [CrossRef]
- Lee, J.H.; Chung, H.C.; Nguyen, V.G.; Moon, H.J.; Kim, H.K.; Park, S.J.; Lee, C.H.; Lee, G.E.; Park, B.K. Detection and phylogenetic analysis of porcine deltacoronavirus in Korean swine farms, 2015. Transbound. Emerg. Dis. 2016, 63, 248–252. [Google Scholar] [CrossRef]
- Marthaler, D.; Raymond, L.; Jiang, Y.; Collins, J.; Rossow, K.; Rovira, A. Rapid detection, complete genome sequencing, and phylogenetic analysis of porcine deltacoronavirus. Emerg. Infect. Dis. 2014, 20, 1347–1350. [Google Scholar] [CrossRef]
- Pérez-Rivera, C.; Ramírez-Mendoza, H.; Mendoza-Elvira, S.; Segura-Velázquez, R.; Sánchez-Betancourt, J.I. First report and phylogenetic analysis of porcine deltacoronavirus in Mexico. Transbound. Emerg. Dis. 2019, 66, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, H.Y. Porcine enteric coronaviruses: An updated overview of the pathogenesis, prevalence, and diagnosis. Vet. Res. Commun. 2021, 45, 75–86. [Google Scholar] [CrossRef]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Elbadry, M.A.; Alam, M.M.; Stephenson, C.J.; Bonny, T.S.; Loeb, J.C.; Telisma, T.; Chavannes, S.; et al. Independent infections of porcine deltacoronavirus among Haitian children. Nature 2021, 600, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wen, Y.; Yu, E.; Yang, J.; Liang, Y.; Song, D.; Wen, Y.; Wu, R.; Zhao, Q.; Du, S.; et al. Identification of an immunodominant neutralizing epitope of porcine deltacoronavirus spike protein. Int. J. Biol. Macromol. 2023, 242 Pt 4, 125190. [Google Scholar] [CrossRef]
- Chen, R.; Fu, J.; Hu, J.; Li, C.; Zhao, Y.; Qu, H.; Wen, X.; Cao, S.; Wen, Y.; Wu, R.; et al. Identification of the immunodominant neutralizing regions in the spike glycoprotein of porcine deltacoronavirus. Virus Res. 2020, 276, 197834. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hulswit, R.J.G.; Kenney, S.P.; Widjaja, I.; Jung, K.; Alhamo, M.A.; van Dieren, B.; van Kuppeveld, F.J.M.; Saif, L.J.; Bosch, B.J. Broad receptor engagement of an emerging global coronavirus may potentiate its diverse cross-species transmissibility. Proc. Natl. Acad. Sci. USA 2018, 115, E5135–E5143. [Google Scholar] [CrossRef]
- Ji, W.; Peng, Q.; Fang, X.; Li, Z.; Li, Y.; Xu, C.; Zhao, S.; Li, J.; Chen, R.; Mo, G.; et al. Structures of a deltacoronavirus spike protein bound to porcine and human receptors. Nat. Commun. 2022, 13, 1467. [Google Scholar] [CrossRef]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef]
- Lee, S.; Lee, C. Functional characterization and proteomic analysis of the nucleocapsid protein of porcine deltacoronavirus. Virus Res. 2015, 208, 136–145. [Google Scholar] [CrossRef]
- Wu, H.; Li, C.; Sun, X.; Cheng, Y.; Chen, Z. Identification of a monoclonal antibody against porcine deltacoronavirus membrane protein. Int. J. Mol. Sci. 2023, 24, 13934. [Google Scholar] [CrossRef]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef]
- Zhou, H.; Shi, K.; Long, F.; Zhao, K.; Feng, S.; Yin, Y.; Xiong, C.; Qu, S.; Lu, W.; Li, Z. A quadruplex qRT-PCR for differential detection of four porcine enteric coronaviruses. Vet. Sci. 2022, 9, 634. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, J.; Liu, H.; Cai, J.; Lin, Q.; Xu, C.; Ding, C.; Liao, M.; Ren, T.; Xiang, B. Phylodynamic analyses of class I Newcastle disease virus isolated in China. Transbound. Emerg. Dis. 2021, 68, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, F.C.; Wu, C.N.; Lin, M.Y.; Hsu, F.Y.; Lin, C.F.; Chang, H.W.; Lin, J.H.; Liu, H.F.; Chiou, M.T.; Chan, K.R.; et al. Phylodynamic analysis and spike protein mutations in porcine deltacoronavirus with a new variant introduction in Taiwan. Virus Evol. 2021, 7, veab096. [Google Scholar] [CrossRef]
- Li, G.; Zhai, S.L.; Zhou, X.; Chen, T.B.; Niu, J.W.; Xie, Y.S.; Si, G.B.; Cong, F.; Chen, R.A.; He, D.S. Phylogeography and evolutionary dynamics analysis of porcine deltacoronavirus with host expansion to humans. Transbound. Emerg. Dis. 2022, 69, e1670–e1681. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yin, Y.; Wang, W.; Cao, L.; Sun, W.; Shi, K.; Lu, H.; Jin, N. Emergence of Thailand-like strains of porcine deltacoronavirus in Guangxi province, China. Vet. Med. Sci. 2020, 6, 854–859. [Google Scholar] [CrossRef]
- He, W.T.; Ji, X.; He, W.; Dellicour, S.; Wang, S.; Li, G.; Zhang, L.; Gilbert, M.; Zhu, H.; Xing, G.; et al. Genomic epidemiology, evolution, and transmission dynamics of porcine deltacoronavirus. Mol. Biol. Evol. 2020, 37, 2641–2654. [Google Scholar] [CrossRef]
- Liu, B.J.; Zuo, Y.Z.; Gu, W.Y.; Luo, S.X.; Shi, Q.K.; Hou, L.S.; Zhong, F.; Fan, J.H. Isolation and phylogenetic analysis of porcine deltacoronavirus from pigs with diarrhoea in Hebei province, China. Transbound. Emerg. Dis. 2018, 65, 874–882. [Google Scholar] [CrossRef]
- Sun, W.; Wang, L.; Huang, H.; Wang, W.; Cao, L.; Zhang, J.; Zheng, M.; Lu, H. Genetic characterization and phylogenetic analysis of porcine deltacoronavirus (PDCoV) in Shandong province, China. Virus Res. 2020, 278, 197869. [Google Scholar] [CrossRef]
- Wang, Z.; Qu, K.; Li, J.; Wang, Y.; Wang, L.; Yu, Y. Prevalence and potential risk factors of PDCoV in pigs based on publications during 2015–2021 in China: Comprehensive literature review and meta-analysis. Microb. Pathog. 2023, 179, 106118. [Google Scholar] [CrossRef]
- Gao, L.; Sun, X.; Yang, H.; Xu, Q.; Li, J.; Kang, J.; Liu, P.; Zhang, Y.; Wang, Y.; Huang, B. Epidemic situation and control measures of African swine fever outbreaks in China 2018–2020. Transbound. Emerg. Dis. 2021, 68, 2676–2686. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Qi, W.; Yang, Y.; Liu, Z.; An, T.; Wu, X.; Chen, J. Prevention and control strategies of African swine fever and progress on pig farm repopulation in China. Viruses 2021, 13, 2552. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, S.; Zhang, B.; Huang, J.; Peng, Q.; Xiao, L.; Yuan, X.; Guo, R.; Zhou, J.; Fan, B.; et al. A novel recombinant S-based subunit vaccine induces protective immunity against porcine deltacoronavirus challenge in piglets. J. Virol. 2023, 97, e0095823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cheng, Y.; Xing, G.; Yu, J.; Liao, A.; Du, L.; Lei, J.; Lian, X.; Zhou, J.; Gu, J. Detection and spike gene characterization in porcine deltacoronavirus in China during 2016–2018. Infect. Genet. Evol. 2019, 73, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, F.C.; Hsu, F.Y.; Chen, Y.H.; Shih, H.C.; Lin, W.H.; Yang, C.Y.; Lin, C.F.; Chiou, M.T.; Lin, C.N. Phylogenetic classification of global porcine deltacoronavirus (PDCoV) reference strains and molecular characterization of PDCoV in Taiwan. Viruses 2021, 13, 1337. [Google Scholar] [CrossRef] [PubMed]
- Amoutzias, G.D.; Nikolaidis, M.; Tryfonopoulou, E.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G. The remarkable evolutionary plasticity of coronaviruses by mutation and recombination: Insights for the COVID-19 pandemic and the future evolutionary paths of SARS-CoV-2. Viruses 2022, 14, 78. [Google Scholar] [CrossRef] [PubMed]
- Salamat, S.E.A.; Collantes, T.M.A.; Lumbera, W.M.L.; Tablizo, F.A.; Mutia, C.T.M.; Ong, J.D.P.; Bandoy, D.J.D.R. Sequence analysis of new variants of porcine epidemic diarrhea virus in Luzon, Philippines, in 2017. Arch. Virol. 2021, 166, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Bahoussi, A.N.; Wang, P.H.; Shah, P.T.; Bu, H.; Wu, C.; Xing, L. Evolutionary plasticity of zoonotic porcine deltacoronavirus (PDCoV): Genetic characteristics and geographic distribution. BMC Vet. Res. 2022, 18, 444. [Google Scholar] [CrossRef] [PubMed]
- Stott, C.J.; Sawattrakool, K.; Saeng-Chuto, K.; Tantituvanont, A.; Nilubol, D. The phylodynamics of emerging porcine deltacoronavirus in Southeast Asia. Transbound. Emerg. Dis. 2022, 69, 2816–2827. [Google Scholar] [CrossRef]
- Guo, X.; Hu, H.; Chen, F.; Li, Z.; Ye, S.; Cheng, S.; Zhang, M.; He, Q. iTRAQ-based comparative proteomic analysis of Vero cells infected with virulent and CV777 vaccine strain-like strains of porcine epidemic diarrhea virus. J. Proteom. 2016, 130, 65–75. [Google Scholar] [CrossRef]
- Zhao, Y.; Qu, H.; Hu, J.; Fu, J.; Chen, R.; Li, C.; Cao, S.; Wen, Y.; Wu, R.; Zhao, Q.; et al. Characterization and pathogenicity of the porcine deltacoronavirus isolated in southwest China. Viruses 2019, 11, 1074. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence (5′→3′) | Product/bp |
|---|---|---|---|
| S | PDCoV-S1-F PDCoV-S1-R PDCoV-S2-F PDCoV-S2-R PDCoV-S3-F PDCoV-S3-R PDCoV-S4-F PDCoV-S4-R PDCoV-S5-F PDCoV-S5-R | CTCGGCTCGTGAGTTAGAGAAG GACCCCGATACAACCTAACATAG CTTCCACCTGATTTAACTGAC GGGTTTTGCCAGTGGTTATA ATGGCATAGCATTCACCTCTC TGCCATTTTCTCAGCATCAAC AATGGCATCATGGTTCTAC CGCTGCAATCAGTTAATTG CGGCAAGCTGATTTCATAC CGCCAAGCGCCCATATGATC | 763 865 824 853 823 |
| M | PDCoV- M -F PDCoV- M -R | AACCCCGTACCTGAGGATGAG GCCGCTTATTGCACAATCTA | 766 |
| N | PDCoV-N1-F PDCoV-N1-R PDCoV-N2-F PDCoV-N2-R | CCATCGCTCCAAGTCATTC CGCCTGAAAGTTGCTCTCA CAAGCTCCCAAGCGGACTTTAC CCAGCCCTAGTTTGCATGATAG | 821 588 |
| Gene | China 1.1 Lineage | China 1.2 Lineage | China 1.3 Lineage |
|---|---|---|---|
| S | 96.6–100.0% | 97.9–100.0% | 96.2–99.9% |
| Gene | Mean Evolutionary Rate (Substitutions/Site/Year) | 95% HPD Interval |
|---|---|---|
| S | 1.907 × 10−3 | 1.5294 × 10−3–2.3232 × 10−3 |
| M | 8.321 × 10−4 | 6.0069 × 10−4–1.0886 × 10−3 |
| N | 1.135 × 10−3 | 7.3867 × 10−4–1.5594 × 10−3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Gao, Y.; Ma, Y.; Shi, K.; Shi, Y.; Feng, S.; Yin, Y.; Long, F.; Sun, W. Genetic and Evolutionary Analysis of Porcine Deltacoronavirus in Guangxi Province, Southern China, from 2020 to 2023. Microorganisms 2024, 12, 416. https://doi.org/10.3390/microorganisms12020416
Li B, Gao Y, Ma Y, Shi K, Shi Y, Feng S, Yin Y, Long F, Sun W. Genetic and Evolutionary Analysis of Porcine Deltacoronavirus in Guangxi Province, Southern China, from 2020 to 2023. Microorganisms. 2024; 12(2):416. https://doi.org/10.3390/microorganisms12020416
Chicago/Turabian StyleLi, Biao, Yeheng Gao, Yan Ma, Kaichuang Shi, Yuwen Shi, Shuping Feng, Yanwen Yin, Feng Long, and Wenchao Sun. 2024. "Genetic and Evolutionary Analysis of Porcine Deltacoronavirus in Guangxi Province, Southern China, from 2020 to 2023" Microorganisms 12, no. 2: 416. https://doi.org/10.3390/microorganisms12020416
APA StyleLi, B., Gao, Y., Ma, Y., Shi, K., Shi, Y., Feng, S., Yin, Y., Long, F., & Sun, W. (2024). Genetic and Evolutionary Analysis of Porcine Deltacoronavirus in Guangxi Province, Southern China, from 2020 to 2023. Microorganisms, 12(2), 416. https://doi.org/10.3390/microorganisms12020416