
Integrated Analysis of the Transcriptome and Microbial Diversity in the Intestine of Miniature Pig Obesity Model
 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Establishment of the Obesity Model
2.2. Material Collection
2.3. Measurement of Body Parameters
2.4. Measurement of Blood Physiological and Biochemical Indicators
2.5. Assessment of Tissue Pathology
2.6. Transcriptome Sequencing
2.7. Validation Using qRT-PCR
2.8. Microbial Diversity Analysis
2.9. Integrated Analysis of Microbial Diversity with Transcriptomics and Physiological Biochemical Indicators
2.10. Statistical Analysis
3. Results
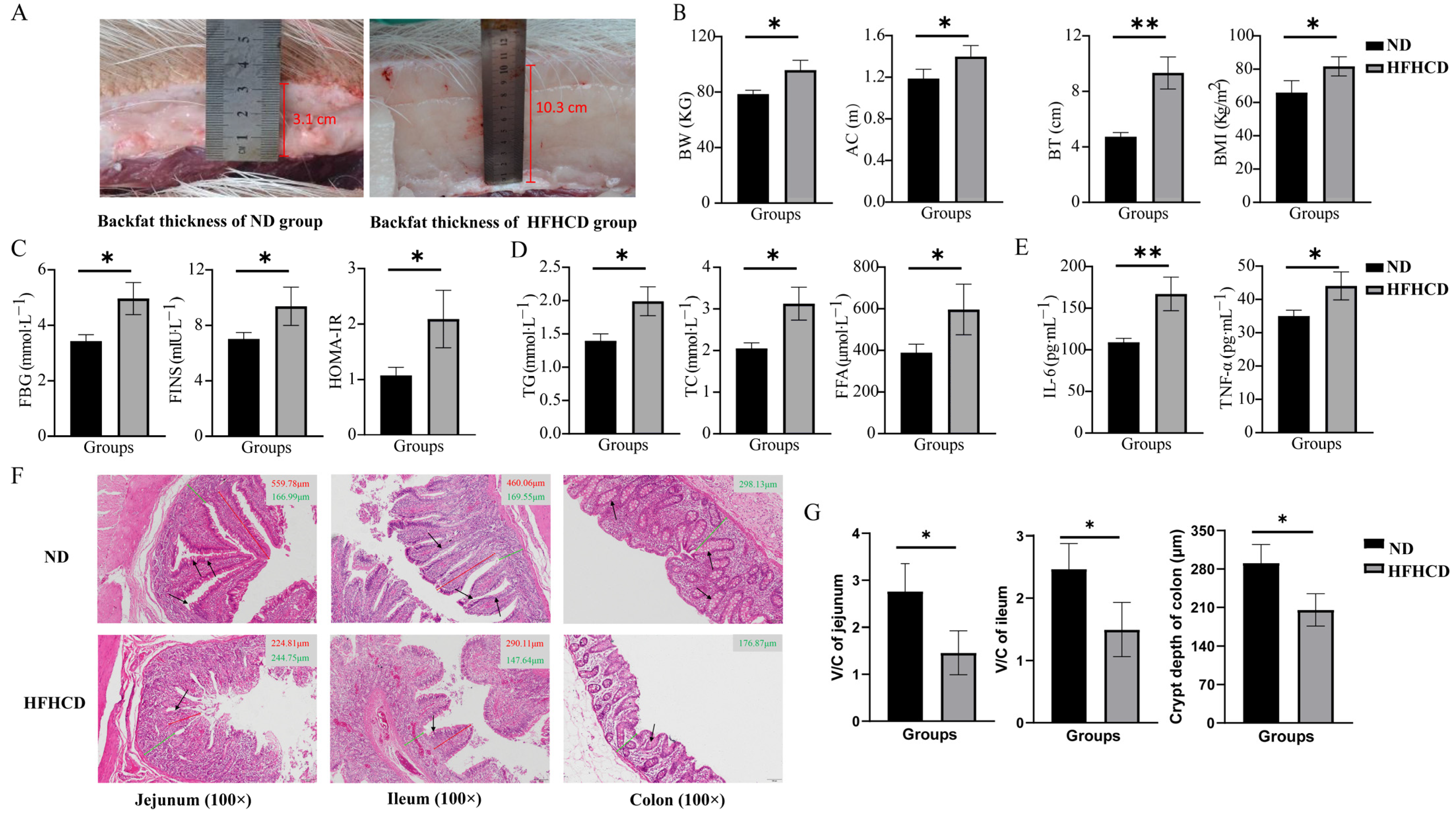
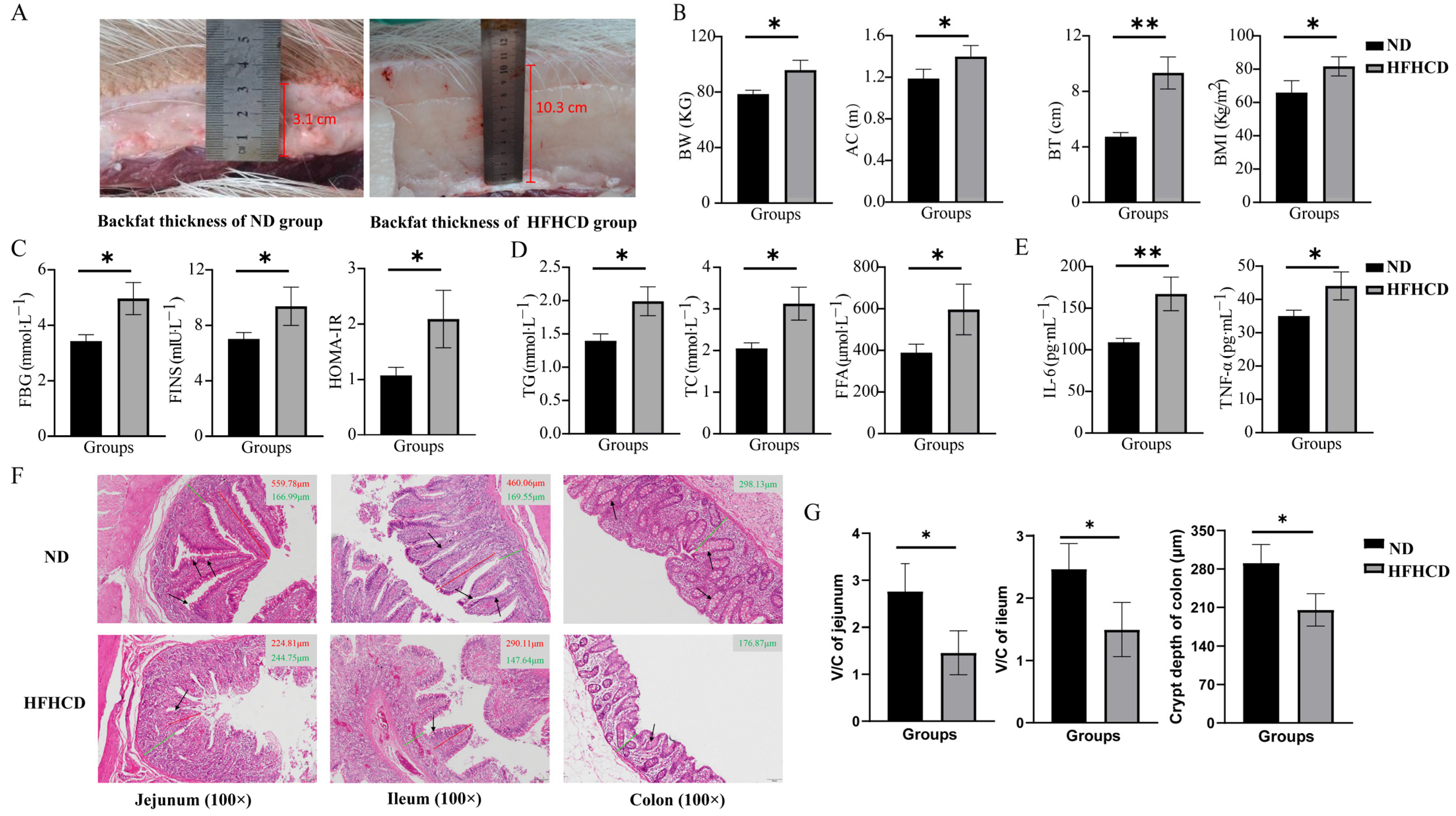
3.1. HFHCD Induced an Elevation in the Obesity Index
3.2. HFHCD-Induced Dyslipidemia, Hyperglycemia, and Inflammatory Response
3.3. Obesity-Impaired Gut Health
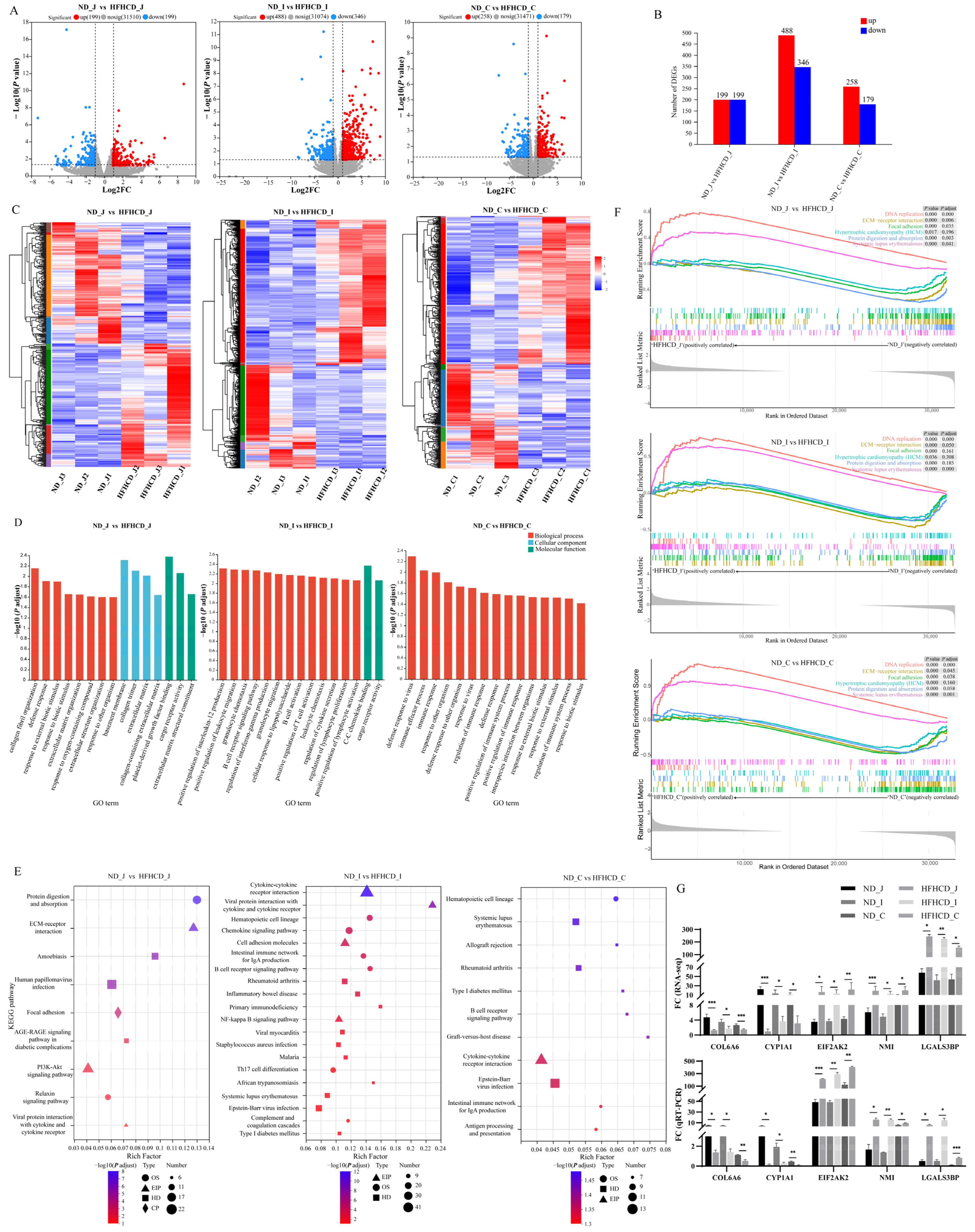
3.4. Transcriptomics and Validation
3.4.1. Identification of Differentially Expressed Genes
3.4.2. Quantitative RT-PCR Verification
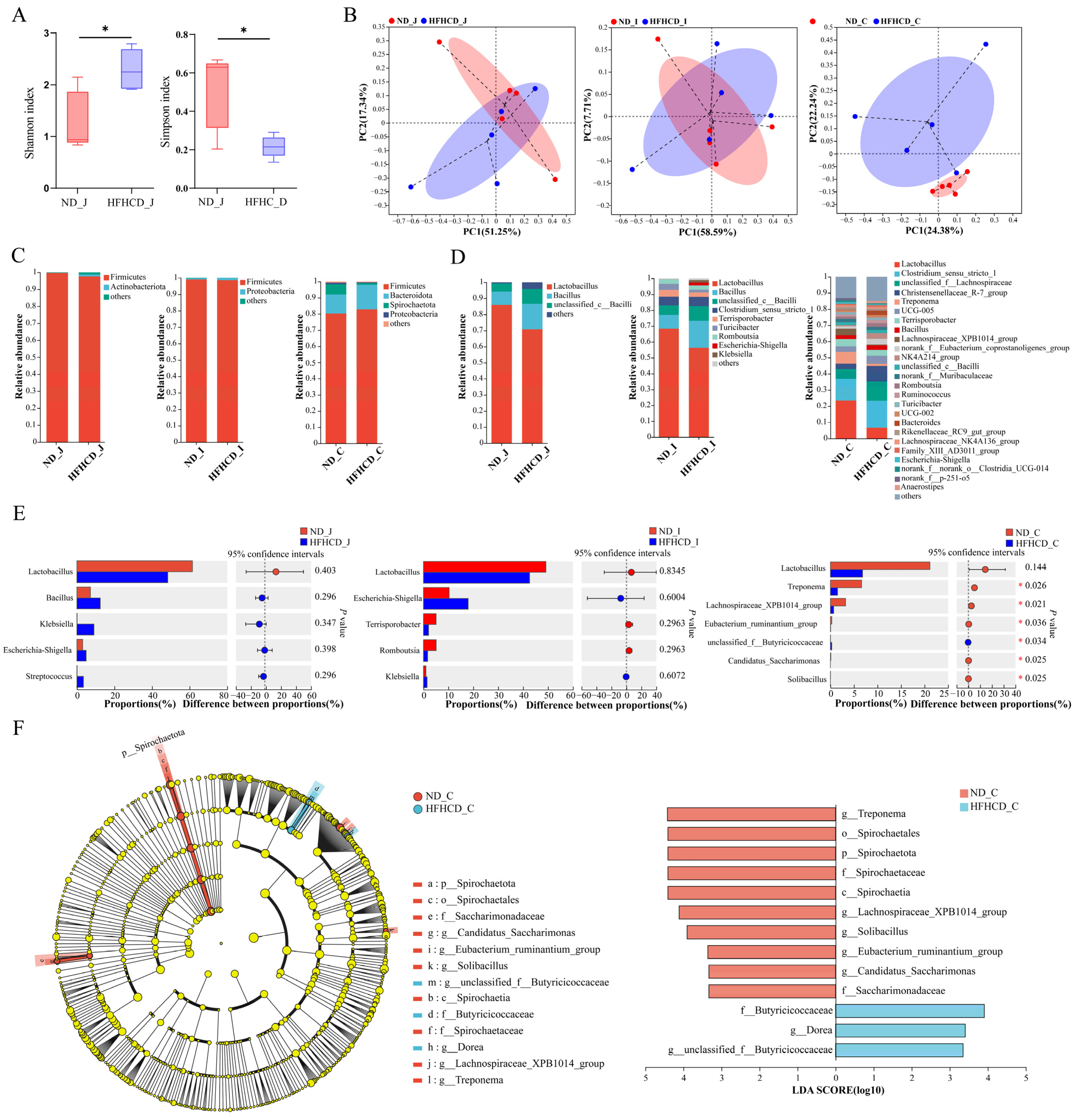
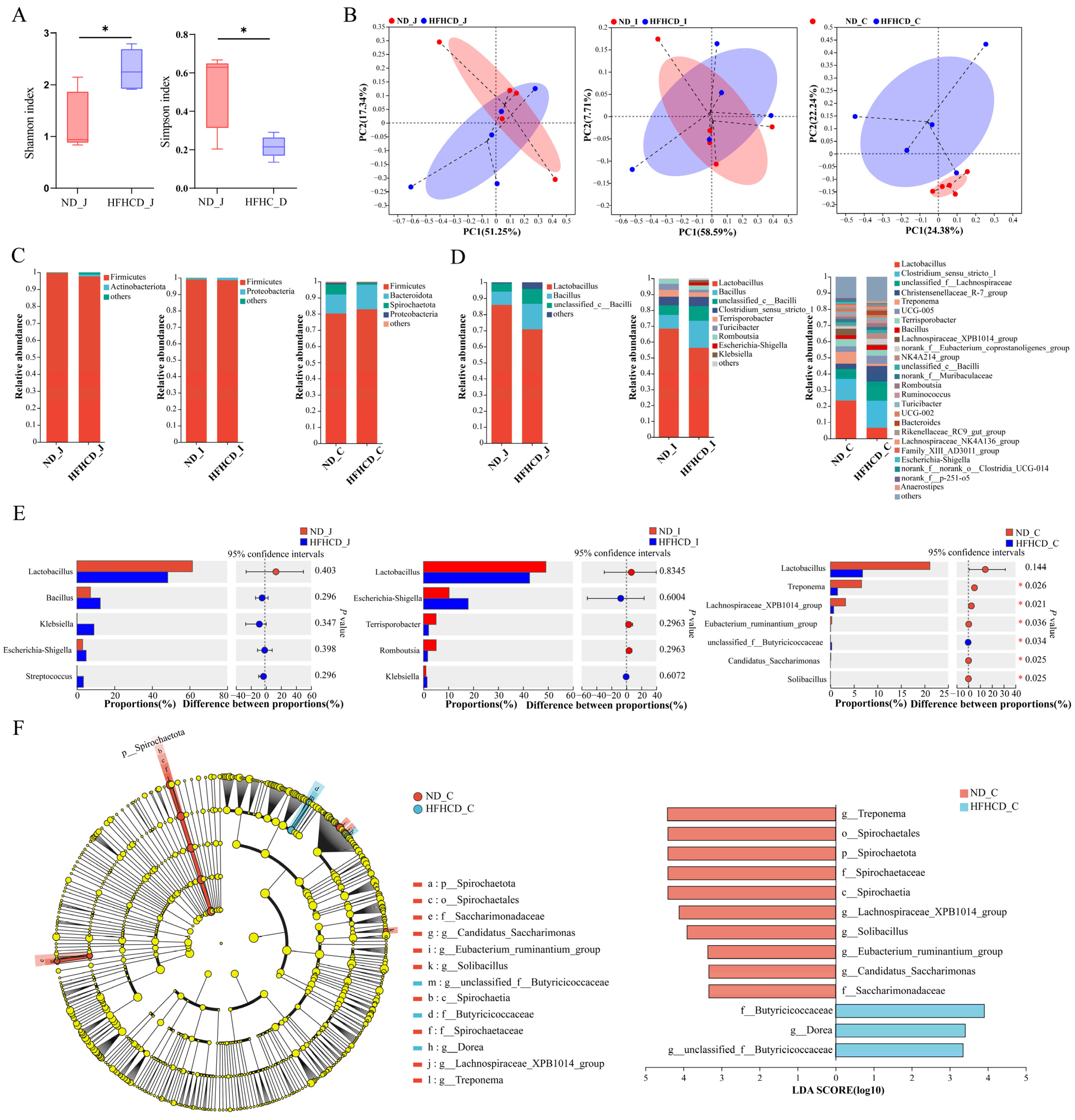
3.5. Microbial Diversity Analysis
3.5.1. Sample Sequence Information
3.5.2. Diversity Analysis
3.5.3. Community Composition and Variation
3.5.4. Differences in Species Composition
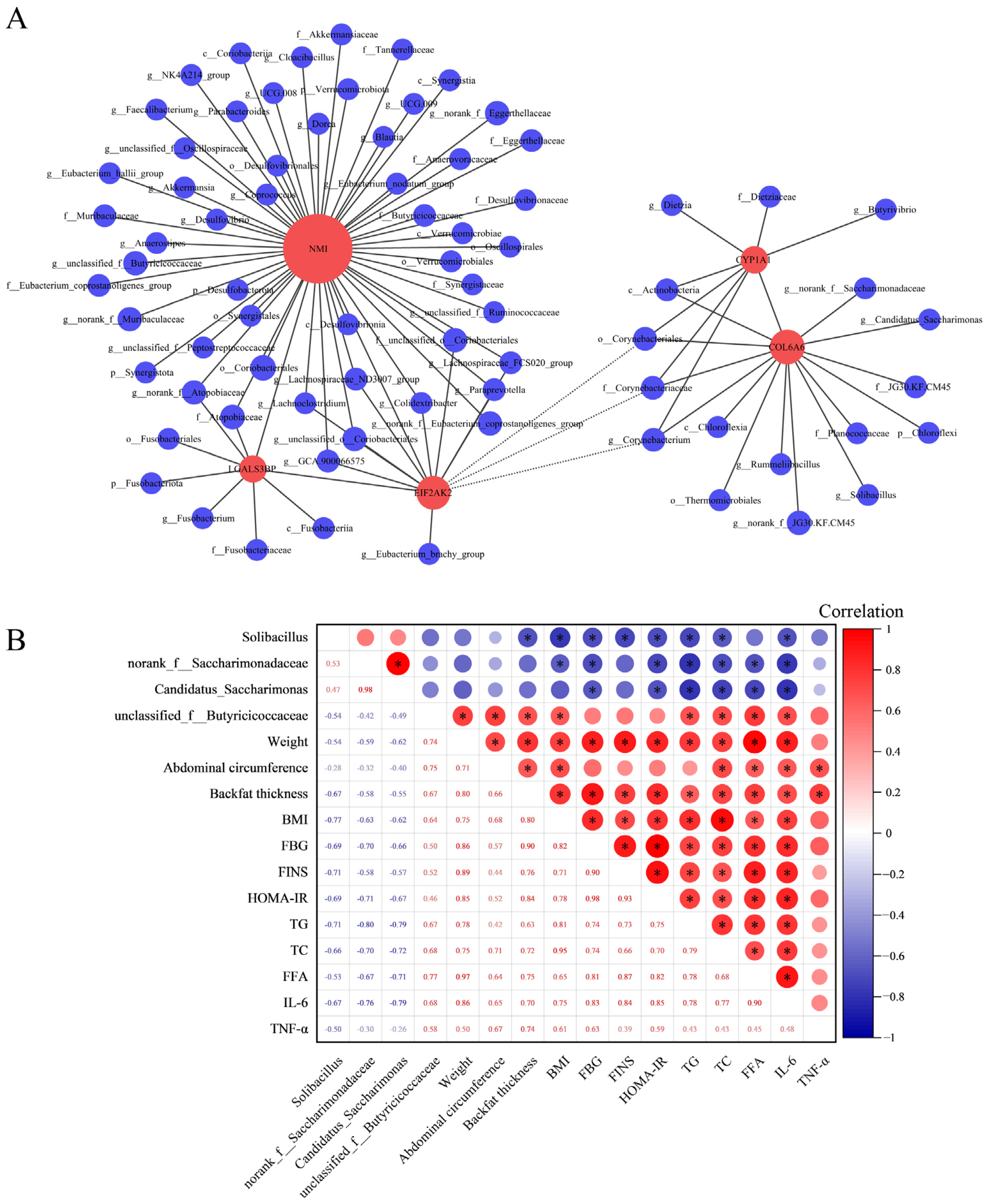
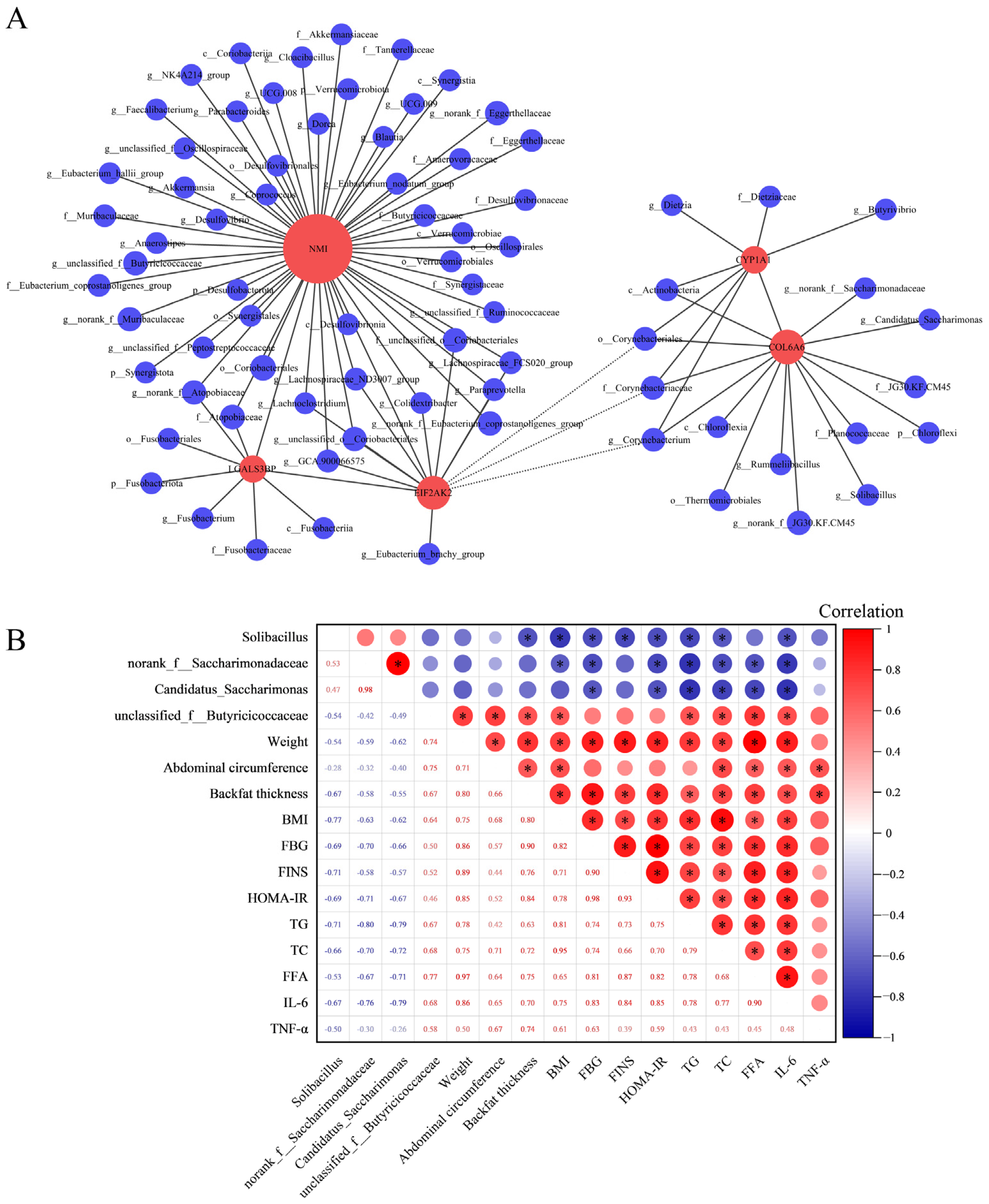
3.6. Integrated Analysis of Microbial Diversity with Transcriptomics and Physiological Biochemical Indicators
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blüher, M. Obesity: Global Epidemiology and Pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Gasmi, A.; Mujawdiya, P.K.; Pivina, L.; Doşa, A.; Semenova, Y.; Benahmed, A.G.; Bjørklund, G. Relationship between Gut Microbiota, Gut Hyperpermeability and Obesity. Curr. Med. Chem. 2021, 28, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Tall, A.R.; Daemen, M.J.A.P.; Falk, E.; Fisher, E.A.; García-Cardeña, G.; Lusis, A.J.; Owens, A.P.; Rosenfeld, M.E.; Virmani, R.; et al. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement from the American Heart Association. Circ. Res. 2017, 121, e53–e79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, Y.; Wang, M.; Guo, Y.; Liang, J.; Yang, X.; Qi, W.; Wu, Y.; Si, J.; Zhu, S.; et al. Development and Genome Sequencing of a Laboratory-Inbred Miniature Pig Facilitates Study of Human Diabetic Disease. iScience 2019, 19, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Si, J.; Zhong, F.; Wu, Y.; Jiang, Q.; Guo, Y.; Yang, X.; Liang, J.; Lan, G. Comparative Analysis of the Transcriptome of T2DM Bama Mini-Pigs with T2DM Patients. Int. J. Diabetes Dev. Ctries. 2022, 42, 236–244. [Google Scholar] [CrossRef]
- Yan, X.; Wu, Y.; Zhong, F.; Jiang, Q.; Zhou, T.; Guo, Y.; Yang, X.; Liang, J.; Joshua Liao, D.; Lan, G. iTRAQ and PRM-Based Quantitative Proteomics in T2DM-Susceptible and -Tolerant Models of Bama Mini-Pig. Gene 2018, 675, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, Y.; Si, J.; Wu, Y.; Wang, M.; Jiang, Q.; Guo, Y.; Liang, J.; Lan, G. Comprehensive Inbred Variation Discovery in Bama Pigs Using de Novo Assemblies. Gene 2018, 679, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome Profiling in Human Diseases: New Advances and Perspectives. Int. J. Mol. Sci. 2017, 18, 1652. [Google Scholar] [CrossRef] [PubMed]
- Zolnikova, O.; Dzhakhaya, N.; Bueverova, E.; Sedova, A.; Kurbatova, A.; Kryuchkova, K.; Butkova, T.; Izotov, A.; Kulikova, L.; Yurku, K.; et al. The Contribution of the Intestinal Microbiota to the Celiac Disease Pathogenesis along with the Effectiveness of Probiotic Therapy. Microorganisms 2023, 11, 2848. [Google Scholar] [CrossRef]
- Angelakis, E.; Merhej, V.; Raoult, D. Related Actions of Probiotics and Antibiotics on Gut Microbiota and Weight Modification. Lancet Infect. Dis. 2013, 13, 889–899. [Google Scholar] [CrossRef]
- Keller, M.; Hopp, L.; Liu, X.; Wohland, T.; Rohde, K.; Cancello, R.; Klös, M.; Bacos, K.; Kern, M.; Eichelmann, F.; et al. Genome-Wide DNA Promoter Methylation and Transcriptome Analysis in Human Adipose Tissue Unravels Novel Candidate Genes for Obesity. Mol. Metab. 2017, 6, 86–100. [Google Scholar] [CrossRef]
- Kang, Y.; Li, Y.; Du, Y.; Guo, L.; Chen, M.; Huang, X.; Yang, F.; Hong, J.; Kong, X. Konjaku Flour Reduces Obesity in Mice by Modulating the Composition of the Gut Microbiota. Int. J. Obes. 2019, 43, 1631–1643. [Google Scholar] [CrossRef]
- Tsai, F.; Coyle, W.J. The Microbiome and Obesity: Is Obesity Linked to Our Gut Flora? Curr. Gastroenterol. Rep. 2009, 11, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ji, J.; Peng, S.; Zhang, Z.; Fang, S.; Li, L.; Zhu, Y.; Huang, L.; Chen, C.; Ma, J. A GWA Study Reveals Genetic Loci for Body Conformation Traits in Chinese Laiwu Pigs and Its Implications for Human BMI. Mamm. Genome 2016, 27, 610–621. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis Model Assessment: Insulin Resistance and Beta-Cell Function from Fasting Plasma Glucose and Insulin Concentrations in Man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A Web Server for Annotation and Identification of Enriched Pathways and Diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, L.; Liang, J.; Jiang, Q.; Guo, Y.; Lan, G. Comparative Analysis on Liver Transcriptome Profiles of Different Methods to Establish Type 2 Diabetes Mellitus Models in Guangxi Bama Mini-Pig. Gene 2018, 673, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Curtasu, M.V.; Knudsen, K.E.B.; Callesen, H.; Purup, S.; Stagsted, J.; Hedemann, M.S. Obesity Development in a Miniature Yucatan Pig Model: A Multi-Compartmental Metabolomics Study on Cloned and Normal Pigs Fed Restricted or Ad Libitum High-Energy Diets. J. Proteome Res. 2019, 18, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Newell-Fugate, A.E.; Taibl, J.N.; Alloosh, M.; Sturek, M.; Bahr, J.M.; Nowak, R.A.; Krisher, R.L. Effects of Obesity and Metabolic Syndrome on Steroidogenesis and Folliculogenesis in the Female Ossabaw Mini-Pig. PLoS ONE 2015, 10, e0128749. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ye, S. Rapamycin Improves Insulin Resistance and Hepatic Steatosis in Type 2 Diabetes Rats through Activation of Autophagy. Cell Biol. Int. 2018, 42, 1282–1291. [Google Scholar] [CrossRef]
- Koleva-Georgieva, D.N.; Sivkova, N.P.; Terzieva, D. Serum Inflammatory Cytokines IL-1beta, IL-6, TNF-Alpha and VEGF Have Influence on the Development of Diabetic Retinopathy. Folia Med. (Plovdiv) 2011, 53, 44–50. [Google Scholar]
- Liu, L.; Wu, C.; Chen, D.; Yu, B.; Huang, Z.; Luo, Y.; Zheng, P.; Mao, X.; Yu, J.; Luo, J.; et al. Selenium-Enriched Yeast Alleviates Oxidative Stress-Induced Intestinal Mucosa Disruption in Weaned Pigs. Oxidative Med. Cell. Longev. 2020, 2020, e5490743. [Google Scholar] [CrossRef]
- Zhang, B.; Fan, X.; Du, H.; Zhao, M.; Zhang, Z.; Zhu, R.; He, B.; Zhang, Y.; Li, X.; Li, J.; et al. Foodborne Carbon Dot Exposure Induces Insulin Resistance through Gut Microbiota Dysbiosis and Damaged Intestinal Mucus Layer. ACS Nano 2023, 17, 6081–6094. [Google Scholar] [CrossRef]
- Rathmell, J.C. Obesity, Immunity, and Cancer. New Engl. J. Med. 2021, 384, 1160–1162. [Google Scholar] [CrossRef]
- Liu, X.; Su, J.; Zhou, H.; Zeng, Z.; Li, Z.; Xiao, Z.; Zhao, M. Collagen VI Antibody Reduces Atherosclerosis by Activating Monocyte/Macrophage Polarization in ApoE−/− Mice. Int. Immunopharmacol. 2022, 111, 1091000. [Google Scholar] [CrossRef]
- Manzella, C.; Singhal, M.; Alrefai, W.A.; Saksena, S.; Dudeja, P.K.; Gill, R.K. Serotonin Is an Endogenous Regulator of Intestinal CYP1A1 via AhR. Sci. Rep. 2018, 8, 6103. [Google Scholar] [CrossRef]
- Ge, L.; Zhang, Y.; Zhao, X.; Wang, J.; Zhang, Y.; Wang, Q.; Yu, H.; Zhang, Y.; You, Y. EIF2AK2 Selectively Regulates the Gene Transcription in Immune Response and Histones Associated with Systemic Lupus Erythematosus. Mol. Immunol. 2021, 132, 132–141. [Google Scholar] [CrossRef]
- Zeng, J.; Yang, Z.; Xu, D.; Song, J.; Liu, Y.; Qin, J.; Weng, Z. NMI Functions as Immuno-Regulatory Molecule in Sepsis by Regulating Multiple Signaling Pathways. Inflammation 2023, 47, 60–73. [Google Scholar] [CrossRef]
- Silverman, A.M.; Nakata, R.; Shimada, H.; Sposto, R.; DeClerck, Y.A. A Galectin-3-Dependent Pathway Upregulates Interleukin-6 in the Microenvironment of Human Neuroblastoma. Cancer Res. 2012, 72, 2228–2238. [Google Scholar] [CrossRef] [PubMed]
- Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/20140211/ (accessed on 23 November 2023).
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in Diversity of the Colonic Mucosa Associated Bacterial Microflora in Patients with Active Inflammatory Bowel Disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in Plasma Endotoxin Concentrations and the Expression of Toll-Like Receptors and Suppressor of Cytokine Signaling-3 in Mononuclear Cells After a High-Fat, High-Carbohydrate Meal: Implications for Insulin Resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.-R.; Lee, J.-C.; Lee, H.-Y.; Kim, M.-S.; Whon, T.W.; Lee, M.-S.; Bae, J.-W. An Increase in the Akkermansia Spp. Population Induced by Metformin Treatment Improves Glucose Homeostasis in Diet-Induced Obese Mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-Talk between Akkermansia Muciniphila and Intestinal Epithelium Controls Diet-Induced Obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; François, P.; de Vos, W.M.; et al. Responses of Gut Microbiota and Glucose and Lipid Metabolism to Prebiotics in Genetic Obese and Diet-Induced Leptin-Resistant Mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef] [PubMed]
- Anastasovska, J.; Arora, T.; Canon, G.J.S.; Parkinson, J.R.C.; Touhy, K.; Gibson, G.R.; Nadkarni, N.A.; So, P.-W.; Goldstone, A.P.; Thomas, E.L.; et al. Fermentable Carbohydrate Alters Hypothalamic Neuronal Activity and Protects Against the Obesogenic Environment. Obesity 2012, 20, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Sultan, S.; El-Mowafy, M.; Elgaml, A.; Ahmed, T.A.E.; Hassan, H.; Mottawea, W. Metabolic Influences of Gut Microbiota Dysbiosis on Inflammatory Bowel Disease. Front. Physiol. 2021, 12, 715506. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, W.-D.; Wang, Y.-D. The Relationship Between Gut Microbiota and Inflammatory Diseases: The Role of Macrophages. Front. Microbiol. 2020, 11, 1065. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, R. Combining Richness and Abundance into a Single Diversity Index Using Matrix Analogues of Shannon’s and Simpson’s Indices. Ecography 2006, 29, 525–530. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A Core Gut Microbiome in Obese and Lean Twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef]
- Yang, K.; Niu, J.; Zuo, T.; Sun, Y.; Xu, Z.; Tang, W.; Liu, Q.; Zhang, J.; Ng, E.K.W.; Wong, S.K.H.; et al. Alterations in the Gut Virome in Obesity and Type 2 Diabetes Mellitus. Gastroenterology 2021, 161, 1257–1269.e13. [Google Scholar] [CrossRef]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of the Gut Microbiota Composition between Obese and Non-Obese Individuals in a Japanese Population, as Analyzed by Terminal Restriction Fragment Length Polymorphism and next-Generation Sequencing. BMC Gastroenterol. 2015, 15, 100. [Google Scholar] [CrossRef]
- Salah, M.; Azab, M.; Ramadan, A.; Hanora, A. New Insights on Obesity and Diabetes from Gut Microbiome Alterations in Egyptian Adults. OMICS A J. Integr. Biol. 2019, 23, 477–485. [Google Scholar] [CrossRef]
- Rojas-Tapias, D.F.; Brown, E.M.; Temple, E.R.; Onyekaba, M.A.; Mohamed, A.M.; Duncan, K.; Schirmer, M.; Walker, R.L.; Mayassi, T.; Pierce, K.A.; et al. Inflammation-Associated Nitrate Facilitates Ectopic Colonization of Oral Bacterium Veillonella Parvula in the Intestine. Nat. Microbiol. 2022, 7, 1673–1685. [Google Scholar] [CrossRef]
- Tap, J.; Mondot, S.; Levenez, F.; Pelletier, E.; Caron, C.; Furet, J.-P.; Ugarte, E.; Muñoz-Tamayo, R.; Paslier, D.L.E.; Nalin, R.; et al. Towards the Human Intestinal Microbiota Phylogenetic Core. Environ. Microbiol. 2009, 11, 2574–2584. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhao, Q.; Wei, Y.; Wang, H.; Ga, Y.; Zhang, Y.; Hao, Z. Pingwei San Ameliorates Spleen Deficiency-Induced Diarrhea through Intestinal Barrier Protection and Gut Microbiota Modulation. Antioxidants 2023, 12, 1122. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-Y.; Zeng, Y.; Hu, G.-Y.; Wang, X.-P. [High-throughput sequencing for analysis of structural change of intestinal microbiota in patients with colorectal adenoma]. Nan Fang Yi Ke Da Xue Xue Bao 2017, 37, 1156–1163. [Google Scholar] [PubMed]
- Singh, S.; Sharma, R.K.; Malhotra, S.; Pothuraju, R.; Shandilya, U.K. Lactobacillus Rhamnosus NCDC17 Ameliorates Type-2 Diabetes by Improving Gut Function, Oxidative Stress and Inflammation in High-Fat-Diet Fed and Streptozotocintreated Rats. Benef. Microbes 2017, 8, 243–255. [Google Scholar] [CrossRef]
- Andreasen, A.S.; Larsen, N.; Pedersen-Skovsgaard, T.; Berg, R.M.G.; Møller, K.; Svendsen, K.D.; Jakobsen, M.; Pedersen, B.K. Effects of Lactobacillus Acidophilus NCFM on Insulin Sensitivity and the Systemic Inflammatory Response in Human Subjects. Br. J. Nutr. 2010, 104, 1831–1838. [Google Scholar] [CrossRef]
- Song, W.; Song, C.; Li, L.; Wang, T.; Hu, J.; Zhu, L.; Yue, T. Lactobacillus Alleviated Obesity Induced by High-Fat Diet in Mice. J. Food Sci. 2021, 86, 5439–5451. [Google Scholar] [CrossRef]
- Miranda-Yuquilema, J.; Taboada, J.; Once, V.; Coyago, M.; Briñez, W. Effect of Agroindustrial Waste Substrate Fermented with Lactic Acid Bacteria and Yeast on Changes in the Gut Microbiota of Guinea Pigs. Microorganisms 2024, 12, 133. [Google Scholar] [CrossRef]
- Wu, Z.-L.; Wei, R.; Tan, X.; Yang, D.; Liu, D.; Zhang, J.; Wang, W. Characterization of Gut Microbiota Dysbiosis of Diarrheic Adult Yaks through 16S rRNA Gene Sequences. Front. Vet. Sci. 2022, 9, 946906. [Google Scholar] [CrossRef]
- Ge, H.; Cai, Z.; Chai, J.; Liu, J.; Liu, B.; Yu, Y.; Liu, J.; Zhang, T. Egg White Peptides Ameliorate Dextran Sulfate Sodium-Induced Acute Colitis Symptoms by Inhibiting the Production of pro-Inflammatory Cytokines and Modulation of Gut Microbiota Composition. Food Chem. 2021, 360, 129981. [Google Scholar] [CrossRef]
- Koontanatechanon, A.; Wongphatcharachai, M.; Nonthabenjawan, N.; Jariyahatthakij, P.; Leksrisompong, P.; Srichana, P.; Prasopdee, S.; Roytrakul, S.; Sriyakul, K.; Thitapakorn, V.; et al. The Effects of Increasing Dietary Fat on Serum Lipid Profile and Modification of Gut Microbiome in C57BL/6N Mice. J. Oleo Sci. 2022, 71, 1039–1049. [Google Scholar] [CrossRef]
- Yu, Q.; Yu, F.; Li, Q.; Zhang, J.; Peng, Y.; Wang, X.; Li, T.; Yin, N.; Sun, G.; Ouyang, H.; et al. Anthocyanin-Rich Butterfly Pea Flower Extract Ameliorating Low-Grade Inflammation in a High-Fat-Diet and Lipopolysaccharide-Induced Mouse Model. J. Agric. Food Chem. 2023, 71, 11941–11956. [Google Scholar] [CrossRef]
- Huang, C.; Li, X.; Wu, L.; Wu, G.; Wang, P.; Peng, Y.; Huang, S.; Yang, Z.; Dai, W.; Ge, L.; et al. The Effect of Different Dietary Structure on Gastrointestinal Dysfunction in Children with Cerebral Palsy and Epilepsy Based on Gut Microbiota. Brain Dev. 2021, 43, 192–199. [Google Scholar] [CrossRef]
- Wong, J.M.W.; de Souza, R.; Kendall, C.W.C.; Emam, A.; Jenkins, D.J.A. Colonic Health: Fermentation and Short Chain Fatty Acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the Gut Microbiota in Nutrition and Health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in Lean and Overweight Healthy Subjects. Obesity (Silver Spring) 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, T.F.S.; Grześkowiak, Ł.; Franceschini, S.C.C.; Bressan, J.; Ferreira, C.L.L.F.; Peluzio, M.C.G. Higher Level of Faecal SCFA in Women Correlates with Metabolic Syndrome Risk Factors. Br. J. Nutr. 2013, 109, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Kaiko, G.E.; Ryu, S.H.; Koues, O.I.; Collins, P.L.; Solnica-Krezel, L.; Pearce, E.J.; Pearce, E.L.; Oltz, E.M.; Stappenbeck, T.S. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 2016, 165, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhuang, J.; Chen, Q.; Xu, L.; Yue, X.; Qiao, D. Chronic Exposure to Polyvinyl Chloride Microplastics Induces Liver Injury and Gut Microbiota Dysbiosis Based on the Integration of Liver Transcriptome Profiles and Full-Length 16S rRNA Sequencing Data. Sci. Total Environ. 2022, 839, 155984. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nutritional Ingredient | Guaranteed Analysis (%) |
|---|---|
| Crude protein | ≥16.0 |
| Lysine | ≥0.8 |
| Crude fiber | ≤6.0 |
| Crude ash | ≤7.0 |
| Calcium | 0.6~1.2 |
| Total phosphorus | 0.4~1.0 |
| Sodium chloride | 0.2~0.8 |
| Water | ≤13.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, W.; Zhu, S.; Feng, L.; Liang, J.; Guo, X.; Cheng, F.; Guo, Y.; Lan, G.; Liang, J. Integrated Analysis of the Transcriptome and Microbial Diversity in the Intestine of Miniature Pig Obesity Model. Microorganisms 2024, 12, 369. https://doi.org/10.3390/microorganisms12020369
Qi W, Zhu S, Feng L, Liang J, Guo X, Cheng F, Guo Y, Lan G, Liang J. Integrated Analysis of the Transcriptome and Microbial Diversity in the Intestine of Miniature Pig Obesity Model. Microorganisms. 2024; 12(2):369. https://doi.org/10.3390/microorganisms12020369
Chicago/Turabian StyleQi, Wenjing, Siran Zhu, Lingli Feng, Jinning Liang, Xiaoping Guo, Feng Cheng, Yafen Guo, Ganqiu Lan, and Jing Liang. 2024. "Integrated Analysis of the Transcriptome and Microbial Diversity in the Intestine of Miniature Pig Obesity Model" Microorganisms 12, no. 2: 369. https://doi.org/10.3390/microorganisms12020369
APA StyleQi, W., Zhu, S., Feng, L., Liang, J., Guo, X., Cheng, F., Guo, Y., Lan, G., & Liang, J. (2024). Integrated Analysis of the Transcriptome and Microbial Diversity in the Intestine of Miniature Pig Obesity Model. Microorganisms, 12(2), 369. https://doi.org/10.3390/microorganisms12020369