
Environmental Changes Driving Shifts in the Structure and Functional Properties of the Symbiotic Microbiota of Daphnia

Abstract
1. Introduction
2. Materials and Methods
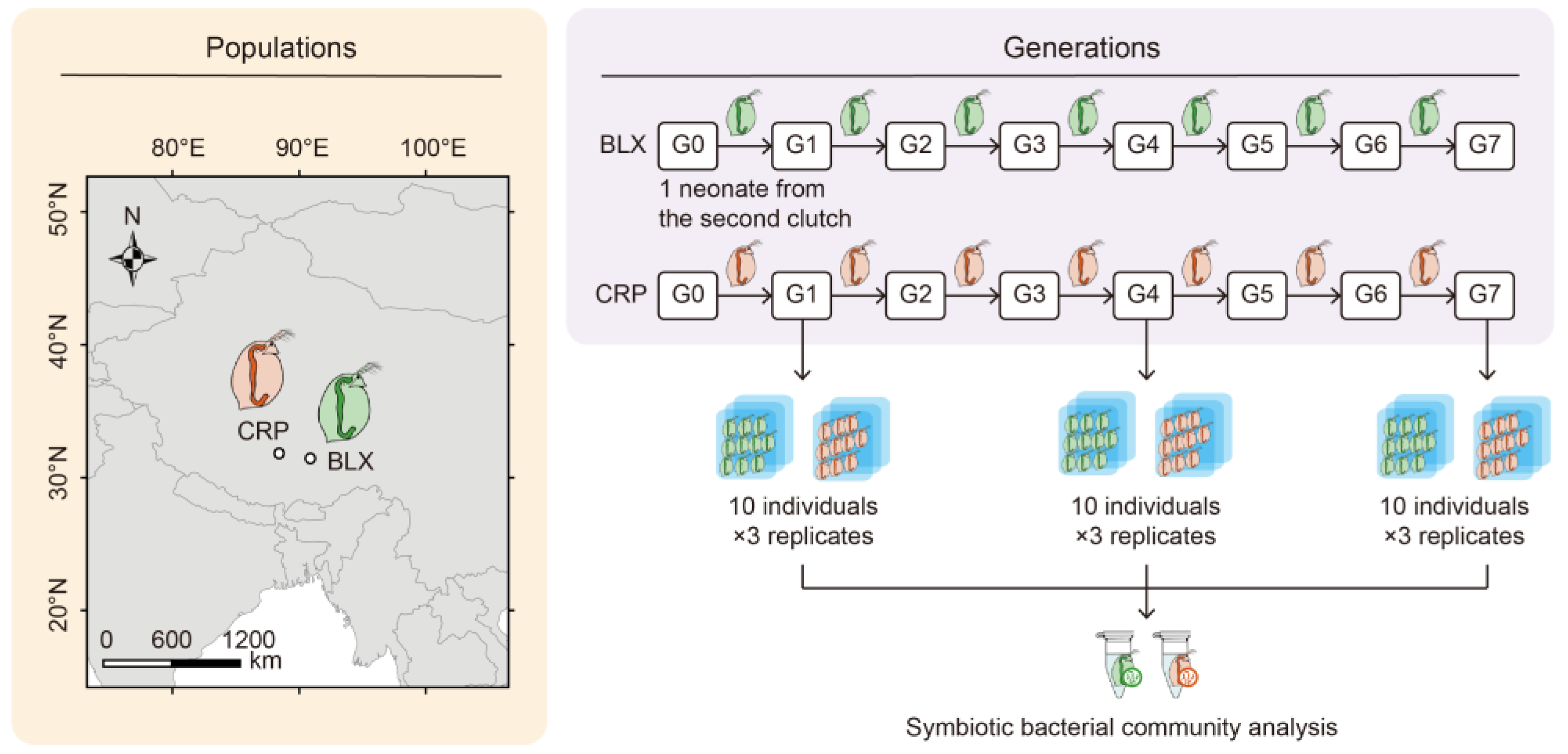
2.1. Research Organism
2.2. Cultivation of Axenic Ankistrodesmus Falcatus
2.3. Experimental Setup
2.4. Microbiota Analysis
3. Results
3.1. Sequencing Results
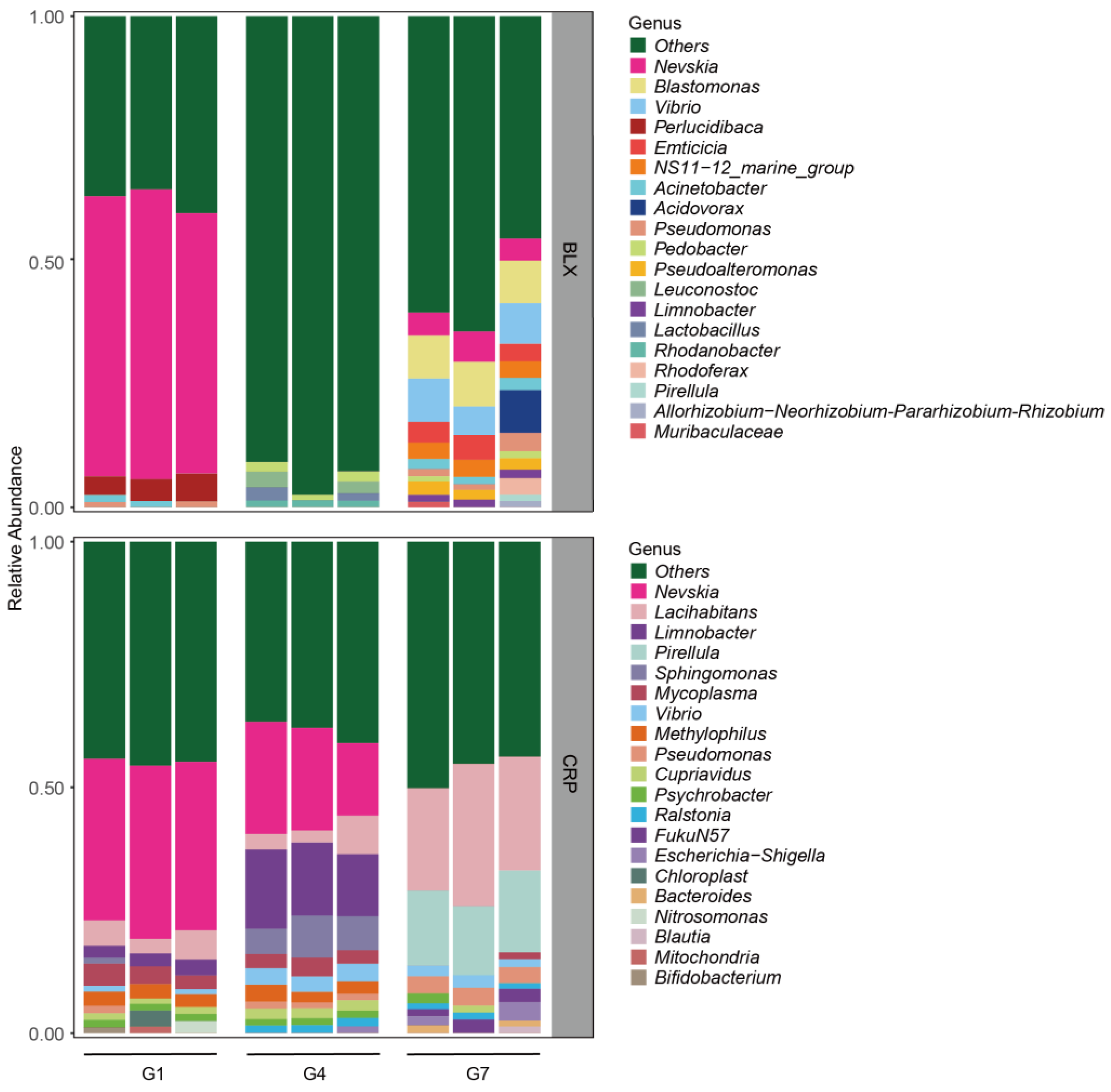
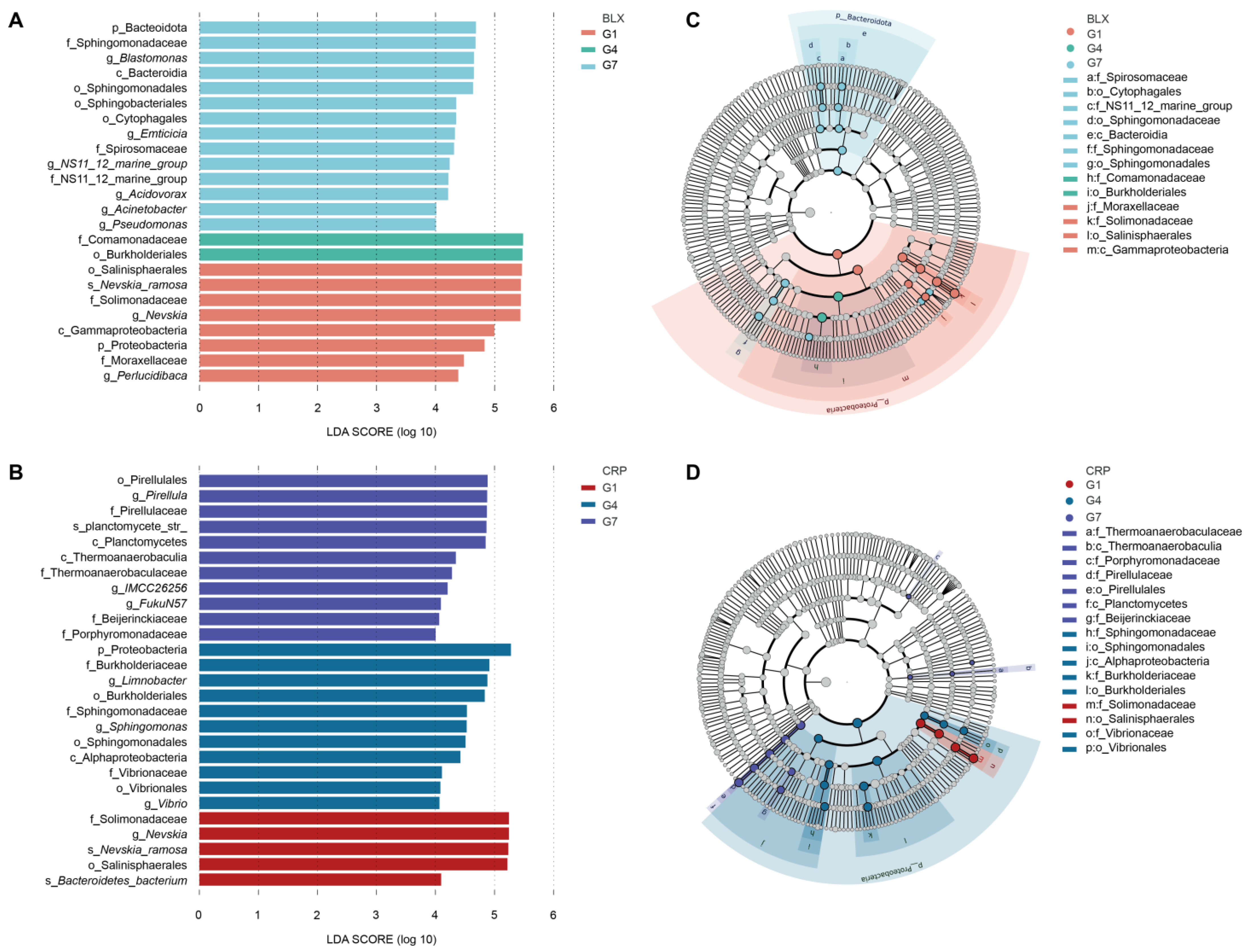
3.2. Changes in Symbiotic Microbiota Across Generations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ezenwa, V.O.; Gerardo, N.M.; Inouye, D.W.; Medina, M.; Xavier, J.B. Animal behavior and the microbiome. Science 2012, 338, 198–199. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Baeckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- McFall-Ngai, M.; Hadfield, M.G.; Bosch, T.C.G.; Carey, H.V.; Domazet-Loso, T.; Douglas, A.E.; Dubilier, N.; Eberl, G.; Fukami, T.; Gilbert, S.F.; et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 2013, 110, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.C.; Kim, S.-H.; You, H.; Kim, B.; Kim, A.C.; Lee, K.-A.; Yoon, J.-H.; Ryu, J.-H.; Lee, W.-J. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 2011, 334, 670–674. [Google Scholar] [CrossRef]
- Sison-Mangus, M.P.; Mushegian, A.A.; Ebert, D. Water fleas require microbiota for survival, growth and reproduction. ISME J. 2015, 9, 59–67. [Google Scholar] [CrossRef]
- Bang, C.; Dagan, T.; Deines, P.; Dubilier, N.; Duschl, W.J.; Fraune, S.; Hentschel, U.; Hirt, H.; Huelter, N.; Lachnit, T.; et al. Metaorganisms in extreme environments: Do microbes play a role in organismal adaptation? Zoology 2018, 127, 1–19. [Google Scholar] [CrossRef]
- Reshef, L.; Koren, O.; Loya, Y.; Zilber-Rosenberg, I.; Rosenberg, E. The coral probiotic hypothesis. Environ. Microbiol. 2006, 8, 2068–2073. [Google Scholar] [CrossRef]
- Kolodny, O.; Schulenburg, H. Microbiome-mediated plasticity directs host evolution along several distinct time scales. Philos. Trans. R. Soc. B-Biol. Sci. 2020, 375, 20190589. [Google Scholar] [CrossRef]
- Baldassarre, L.; Ying, H.; Reitzel, A.; Franzenburgq, S.; Fraune, S. Microbiota mediated plasticity promotes thermal adaptation in the sea anemone Nematostella vectensis. Nat. Commun. 2022, 13, 3804. [Google Scholar] [CrossRef]
- Lampert, W. Daphnia: Development of a Model Organism in Ecology and Evolution. In Excellence in Ecology; Inter Research: Oldendorf/Luhe, Germany, 2011. [Google Scholar]
- Bekker, E.I.; Karabanov, D.P.; Galimov, Y.R.; Haag, C.R.; Neretina, T.V.; Kotov, A.A. Phylogeography of Daphnia magna Straus (Crustacea: Cladocera) in Northern Eurasia: Evidence for a deep longitudinal split between mitochondrial lineages. PLoS ONE 2018, 13, e0194045. [Google Scholar] [CrossRef]
- Fields, P.D.; Reisser, C.; Dukic, M.; Haag, C.R.; Ebert, D. Genes mirror geography in Daphnia magna. Mol. Ecol. 2015, 24, 4521–4536. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ni, Y.; Wang, X.; Hu, W.; Yin, M. Lineage diversity, morphological and genetic divergence in Daphnia magna (Crustacea) among Chinese lakes at different altitudes. Contrib. Zool. 2020, 89, 450–470. [Google Scholar] [CrossRef]
- Peerakietkhajorn, S.; Kato, Y.; Kasalicky, V.; Matsuura, T.; Watanabe, H. Betaproteobacteria limnohabitans strains increase fecundity in the crustacean Daphnia magna: Symbiotic relationship between major bacterioplankton and zooplankton in freshwater ecosystem. Environ. Microbiol. 2016, 18, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Mushegian, A.A.; Walser, J.-C.; Sullam, K.E.; Ebert, D. The microbiota of diapause: How host-microbe associations are formed after dormancy in an aquatic crustacean. J. Anim. Ecol. 2018, 87, 400–413. [Google Scholar] [CrossRef]
- Cooper, R.O.; Cressler, C.E. Characterization of key bacterial species in the Daphnia magna microbiota using shotgun metagenomics. Sci. Rep. 2020, 10, 652. [Google Scholar] [CrossRef]
- Macke, E.; Callens, M.; De Meester, L.; Decaestecker, E. Host-genotype dependent gut microbiota drives zooplankton tolerance to toxic cyanobacteria. Nat. Commun. 2017, 8, 1608. [Google Scholar] [CrossRef]
- Hegg, A.; Radersma, R.; Uller, T. A field experiment reveals seasonal variation in the Daphnia gut microbiome. Oikos 2021, 130, 2191–2201. [Google Scholar] [CrossRef]
- Thompson, L.G.; Yao, T.; Mosley-Thompson, E.; Davis, M.E.; Henderson, K.A.; Lin, P.N. A high-resolution millennial record of the South Asian Monsoon from Himalayan ice cores. Science 2000, 289, 1916–1919. [Google Scholar] [CrossRef]
- Clewing, C.; Albrecht, C.; Wilke, T. A complex system of Glacial sub-Refugia drives endemic freshwater biodiversity on the Tibetan Plateau. PLoS ONE 2016, 11, e0160286. [Google Scholar] [CrossRef]
- Niu, Y.; Yang, S.; Zhou, J.; Chu, B.; Ma, S.; Zhu, H.; Hua, L. Vegetation distribution alongmountain environmental gradient predicts shifts in plant community response to climate change in alpine meadow on the Tibetan Plateau. Sci. Total Environ. 2019, 650, 505–514. [Google Scholar] [CrossRef]
- Folmer, O.F.; Black, M.B.; Hoeh, W.R.; Lutz, R.A.; Vrijenhoek, R.C. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Kilham, S.S.; Kreeger, D.A.; Lynn, S.G.; Goulden, C.E.; Herrera, L. COMBO: A defined freshwater culture medium for algae and zooplankton. Hydrobiologia 1998, 377, 147–159. [Google Scholar] [CrossRef]
- Pichler, M.; Coskun, O.K.; Ortega-Arbulu, A.-S.; Conci, N.; Woerheide, G.; Vargas, S.; Orsi, W.D. A 16S rRNA gene sequencing and analysis protocol for the Illumina MiniSeq platform. Microbiologyopen 2018, 7, e00611. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2 (vol 37, pg 852, 2019). Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Costantini, M.S.; Medeiros, M.C.I.; Crampton, L.H.; Reed, F.A. Wild gut microbiomes reveal individuals, species, and location as drivers of variation in two critically endangered Hawaiian honeycreepers. PeerJ 2021, 9, e12291. [Google Scholar] [CrossRef]
- Locher, K.; Belanger, C.R.; Eckbo, E.; Caza, M.; Velapatino, B.; Charles, M.K. Automated 16S Sequencing Using an R-Based Analysis Module for Bacterial Identification. Microbiol. Spectr. 2022, 10, e0040822. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Webster, N.S.; Reusch, T.B.H. Microbial contributions to the persistence of coral reefs. ISME J. 2017, 11, 2167–2174. [Google Scholar] [CrossRef]
- van Oppen, M.J.H.; Blackall, L.L. Coral microbiome dynamics, functions and design in a changing world. Nat. Rev. Microbiol. 2019, 17, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, A.; Aizpurua, O.; Bohmann, K.; Zepeda-Mendoza, M.L.; Gilbert, M.T.P. Do vertebrate gut metagenomes confer rapid ecological adaptation? Trends Ecol. Evol. 2016, 31, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Nong, G.; Preston, J.F.; Ben-Ami, F.; Ebert, D. Comparative metagenomics of Daphnia symbionts. BMC Genom. 2009, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Callens, M.; Watanabe, H.; Kato, Y.; Miura, J.; Decaestecker, E. Microbiota inoculum composition affects holobiont assembly and host growth in Daphnia. Microbiome 2018, 6, 56. [Google Scholar] [CrossRef]
- Akbar, S.; Huang, J.; Zhou, Q.; Gu, L.; Sun, Y.; Zhang, L.; Lyu, K.; Yang, Z. Elevated temperature and toxic Microcystis reduce Daphnia fitness and modulate gut microbiota. Environ. Pollut. 2021, 271, 116409. [Google Scholar] [CrossRef]
- Peerakietkhajorn, S.; Tsukada, K.; Kato, Y.; Matsuura, T.; Watanabe, H. Symbiotic bacteria contribute to increasing the population size of a freshwater crustacean, Daphnia magna. Environ. Microbiol. Rep. 2015, 7, 364–372. [Google Scholar] [CrossRef]
- Gorokhova, E.; Rivetti, C.; Furuhagen, S.; Edlund, A.; Ek, K.; Breitholtz, M. Bacteria-mediated effects of antibiotics on Daphnia nutrition. Environ. Sci. Technol. 2015, 49, 5779–5787. [Google Scholar] [CrossRef]
- Pladdies, T.; Babenzien, H.D.; Cypionka, H. Distribution of Nevskia ramosa and other rosette-forming neustonic bacteria. Microb. Ecol. 2004, 47, 218–223. [Google Scholar] [CrossRef]
- Sturmeyer, H.; Overmann, J.; Babenzien, H.D.; Cypionka, H. Ecophysiological and phylogenetic studies of Nevskia ramosa in pure culture. Appl. Environ. Microbiol. 1998, 64, 1890–1894. [Google Scholar] [CrossRef]
- Wotton, R.S.; Preston, T.M. Surface films: Areas of water bodies that are often overlooked. Bioscience 2005, 55, 137–145. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, Y.; Wang, H.; Zhang, H.; Chen, W.; Lu, W. Lactic acid bacteria-derived exopolysaccharide: Formation, immunomodulatory ability, health effects, and structure-function relationship. Microbiol. Res. 2023, 274, 127432. [Google Scholar] [CrossRef] [PubMed]
- Zikmanis, P.; Brants, K.; Kolesovs, S.; Semjonovs, P. Extracellular polysaccharides produced by bacteria of the Leuconostoc genus. World J. Microbiol. Biotechnol. 2020, 36, 161. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Singh, A. Gut microbiome and human health: Exploring how the probiotic genus Lactobacillus modulate immune responses. Front. Pharmacol. 2022, 13, 1042189. [Google Scholar] [CrossRef] [PubMed]
- Capurso, L. Thirty Years of Lactobacillus rhamnosus GG: A Review. J. Clin. Gastroenterol. 2019, 53 (Suppl. S1), S1–S41. [Google Scholar] [CrossRef]
- Chiriac, M.-C.; Haber, M.; Salcher, M.M. Adaptive genetic traits in pelagic freshwater microbes. Environ. Microbiol. 2023, 25, 606–641. [Google Scholar] [CrossRef]
- Compant, S.; Nowak, J.; Coenye, T.; Clement, C.; Barka, E.A. Diversity and occurrence of Burkholderia spp. in the natural environment. FEMS Microbiol. Rev. 2008, 32, 607–626. [Google Scholar] [CrossRef]
- Nowak, J.; Shulaev, V. Priming for transplant stress resistance in in vitro propagation. In Vitro Cell. Dev. Biol.-Plant 2003, 39, 107–124. [Google Scholar] [CrossRef]
- O’Sullivan, L.A.; Weightman, A.J.; Jones, T.H.; Marchbank, A.M.; Tiedje, J.M.; Mahenthiralingam, E. Identifying the genetic basis of ecologically and biotechnologically useful functions of the bacterium Burkholderia vietnamiensis. Environ. Microbiol. 2007, 9, 1017–1034. [Google Scholar] [CrossRef]
- Depoorter, E.; Bull, M.J.; Peeters, C.; Coenye, T.; Vandamme, P.; Mahenthiralingam, E. Burkholderia: An update on taxonomy and biotechnological potential as antibiotic producers. Appl. Microbiol. Biotechnol. 2016, 100, 5215–5229. [Google Scholar] [CrossRef]
- Lindstrom, E.S.; Langenheder, S. Local and regional factors influencing bacterial community assembly. Environ. Microbiol. Rep. 2012, 4, 1–9. [Google Scholar] [CrossRef]
- Eckert, E.M.; Anicic, N.; Fontaneto, D. Freshwater zooplankton microbiome composition is highly flexible and strongly influenced by the environment. Mol. Ecol. 2021, 30, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Callens, M.; De Meester, L.; Muylaert, K.; Mukherjee, S.; Decaestecker, E. The bacterioplankton community composition and a host genotype dependent occurrence of taxa shape the Daphnia magna gut bacterial community. FEMS Microbiol. Ecol. 2020, 96, fiaa128. [Google Scholar] [CrossRef] [PubMed]
- Frankel-Bricker, J.; Song, M.J.; Benner, M.J.; Schaack, S. Variation in the microbiota associated with Daphnia magna across genotypes, populations, and temperature. Microb. Ecol. 2020, 79, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Sullam, K.E.; Pichon, S.; Schaer, T.M.M.; Ebert, D. The combined effect of temperature and host clonal line on the microbiota of a planktonic Crustacean. Microb. Ecol. 2018, 76, 506–517. [Google Scholar] [CrossRef]
- Akbar, S.; Li, X.; Ding, Z.; Liu, Q.; Huang, J.; Zhou, Q.; Gu, L.; Yang, Z. Disentangling diet- and medium-associated microbes in shaping Daphnia gut microbiome. Microb. Ecol. 2022, 84, 911–921. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw PE | Combined | Qualified | Nochime | Base (nt) | AvgLen (nt) | Q20 | Q30 | GC% | Effective% |
|---|---|---|---|---|---|---|---|---|---|---|
| BLX-G1.1 | 83,200 | 82,788 | 82,566 | 74,012 | 18,725,658 | 253 | 99.34 | 97.49 | 54.37 | 88.96 |
| BLX-G1.2 | 89,087 | 87,990 | 87,424 | 77,536 | 19,616,285 | 253 | 99.29 | 97.37 | 54.35 | 87.03 |
| BLX-G1.3 | 79,898 | 79,652 | 79,472 | 69,472 | 17,576,179 | 253 | 99.36 | 97.55 | 54.36 | 86.95 |
| BLX-G4.1 | 70,345 | 68,448 | 68,224 | 63,773 | 16,133,089 | 253 | 99.49 | 98.08 | 54.23 | 90.66 |
| BLX-G4.2 | 80,580 | 79,809 | 79,604 | 74,448 | 18,832,339 | 253 | 99.52 | 98.14 | 54.43 | 92.39 |
| BLXG4.3 | 80,695 | 79,482 | 79,208 | 74,430 | 18,824,748 | 253 | 99.5 | 98.08 | 54.29 | 92.24 |
| BLXG7.1 | 87,116 | 85,198 | 84,891 | 61,169 | 15,481,354 | 253 | 99.18 | 97.04 | 53.49 | 70.22 |
| BLXG7.2 | 88,874 | 88,368 | 88,212 | 66,341 | 16,786,801 | 253 | 99.31 | 97.42 | 53.61 | 74.65 |
| BLXG7.3 | 84,465 | 82,614 | 82,317 | 67,282 | 17,026,164 | 253 | 99.29 | 97.38 | 53.48 | 79.66 |
| CRPG-1.1 | 79,628 | 78,796 | 78,717 | 67,911 | 17,189,725 | 253 | 99.61 | 98.43 | 53.44 | 85.29 |
| CRPG1.2 | 87,199 | 86,575 | 86,511 | 75,022 | 18,981,147 | 253 | 99.63 | 98.48 | 53.87 | 86.04 |
| CRPG 1.3 | 79,676 | 79,114 | 79,058 | 67,761 | 17,150,369 | 253 | 99.62 | 98.44 | 53.44 | 85.05 |
| CRP-G4.1 | 82,658 | 81,468 | 81,393 | 71,177 | 18,024,238 | 253 | 99.64 | 98.52 | 53.39 | 86.11 |
| CRP-G4.2 | 79,841 | 78,507 | 78,456 | 65,134 | 16,484,792 | 253 | 99.62 | 98.46 | 53.3 | 81.58 |
| CRP-G4.3 | 84,457 | 82,625 | 82,552 | 72,148 | 18,293,095 | 254 | 99.62 | 98.43 | 53.05 | 85.43 |
| CRP-G7.1 | 73,089 | 71,654 | 71,586 | 65,504 | 16,608,432 | 254 | 99.61 | 98.43 | 52.61 | 89.62 |
| CRP-G7.2 | 85,040 | 83,639 | 83,560 | 74,237 | 18,830,649 | 254 | 99.59 | 98.34 | 52.18 | 87.3 |
| CRP-G7.3 | 84,511 | 83,075 | 83,002 | 71,923 | 18,242,441 | 254 | 99.62 | 98.44 | 52.48 | 85.1 |
| Taxonomy | BLX -G1.1 | BLX -G1.2 | BLX -G1.3 | BLX -G4.1 | BLX -G4.2 | BLX -G4.3 | BLX -G7.1 | BLX -G7.2 | BLX -G7.3 | Tax_Detail |
|---|---|---|---|---|---|---|---|---|---|---|
| Nevskia | 0.57 | 0.59 | 0.53 | 0 | 0 | 0 | 0.05 | 0.06 | 0.04 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Salinisphaerales;f_Solimonadaceae;g_Nevskia; |
| Blastomonas | 0 | 0 | 0 | 0 | 0 | 0 | 0.09 | 0.09 | 0.09 | k_Bacteria;p_Proteobacteria;c_Alphaproteobacteria; o_Sphingomonadales;f_Sphingomonadaceae; g_Blastomonas; |
| Vibrio | 0 | 0 | 0 | 0 | 0 | 0 | 0.09 | 0.06 | 0.08 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Vibrionales;f_Vibrionaceae;g_Vibrio; |
| Acidovorax | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.09 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Burkholderiales;f_Comamonadaceae;g_Acidovorax; |
| Perlucidibaca | 0.04 | 0.04 | 0.06 | 0 | 0 | 0 | 0 | 0 | 0 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Pseudomonadales;f_Moraxellaceae;g_Perlucidibaca; |
| Emticicia | 0 | 0 | 0 | 0 | 0 | 0 | 0.04 | 0.05 | 0.04 | k_Bacteria;p_Bacteroidota;c_Bacteroidia;o_Cytophagales;f_Spirosomaceae;g_Emticicia; |
| Pseudomonas | 0.01 | 0 | 0.01 | 0 | 0 | 0 | 0.01 | 0.01 | 0.04 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Pseudomonadales;f_Pseudomonadaceae; g_Pseudomonas; |
| NS11-12_marine_ group | 0 | 0 | 0 | 0 | 0 | 0 | 0.03 | 0.04 | 0.03 | k_Bacteria;p_Bacteroidota;c_Bacteroidia; o_Sphingobacteriales;f_NS11-12_marine_group;g_NS11-12_marine_group; |
| Rhodoferax | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.03 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Burkholderiales;f_Comamonadaceae;g_Rhodoferax; |
| Leuconostoc | 0 | 0 | 0 | 0.03 | 0 | 0.02 | 0 | 0 | 0 | k_Bacteria;p_Firmicutes;c_Bacilli;o_Lactobacillales; f_Leuconostocaceae;g_Leuconostoc; |
| Pseudoalteromonas | 0 | 0 | 0 | 0 | 0 | 0 | 0.03 | 0.02 | 0.02 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Alteromonadales;f_Pseudoalteromonadaceae; g_Pseudoalteromonas; |
| Lactobacillus | 0 | 0 | 0 | 0.03 | 0 | 0.02 | 0 | 0 | 0 | k_Bacteria;p_Firmicutes;c_Bacilli;o_Lactobacillales; f_Lactobacillaceae;g_Lactobacillus; |
| Acinetobacter | 0.01 | 0.01 | 0 | 0 | 0 | 0 | 0.02 | 0.01 | 0.06 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Pseudomonadales;f_Moraxellaceae;g_Acinetobacter; |
| Pedobacter | 0 | 0 | 0 | 0.02 | 0.01 | 0.02 | 0.01 | 0 | 0.01 | k_Bacteria;p_Bacteroidota;c_Bacteroidia; o_Sphingobacteriales;f_Sphingobacteriaceae;g_Pedobacter; |
| Limnobacter | 0 | 0 | 0 | 0 | 0 | 0 | 0.01 | 0.02 | 0.02 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Burkholderiales;f_Burkholderiaceae;g_Limnobacter; |
| Rhodanobacter | 0 | 0 | 0 | 0.01 | 0.01 | 0.01 | 0 | 0 | 0 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Xanthomonadales;f_Rhodanobacteraceae; g_Rhodanobacter; |
| Pirellula | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.01 | k_Bacteria;p_Planctomycetota;c_Planctomycetes; o_Pirellulales;f_Pirellulaceae;g_Pirellula; |
| Allorhizobium-Neo | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.01 | k_Bacteria;p_Proteobacteria;c_Alphaproteobacteria; o_Rhizobiales;f_Rhizobiaceae; g_Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium; |
| Muribaculaceae | 0 | 0 | 0 | 0 | 0 | 0 | 0.01 | 0 | 0 | k_Bacteria;p_Bacteroidota;c_Bacteroidia;o_Bacteroidales; f_Muribaculaceae;g_Muribaculaceae; |
| Taxonomy | CRP -G1.1 | CRP -G1.2 | CRP -G1.3 | CRP -G4.1 | CRP -G4.2 | CRP -G4.3 | CRP -G7.1 | CRP -G7.2 | CRP -G7.3 | Tax_Detail |
|---|---|---|---|---|---|---|---|---|---|---|
| Nevskia | 0.33 | 0.35 | 0.34 | 0.23 | 0.21 | 0.15 | 0.00 | 0.00 | 0.00 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Salinisphaerales;f_Solimonadaceae;g_Nevskia; |
| Lacihabitans | 0.05 | 0.03 | 0.06 | 0.03 | 0.02 | 0.08 | 0.21 | 0.29 | 0.23 | k_Bacteria;p_Bacteroidota;c_Bacteroidia; o_Cytophagales;f_Spirosomaceae;g_Lacihabitans; |
| Pirellula | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.15 | 0.14 | 0.17 | k_Bacteria;p_Planctomycetota;c_Planctomycetes; o_Pirellulales;f_Pirellulaceae;g_Pirellula; |
| Limnobacter | 0.02 | 0.03 | 0.03 | 0.16 | 0.15 | 0.13 | 0.00 | 0.00 | 0.00 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria; o_Burkholderiales;f_Burkholderiaceae;g_Limnobacter; |
| Sphingomonas | 0.01 | 0.00 | 0.00 | 0.05 | 0.09 | 0.07 | 0.00 | 0.00 | 0.00 | k_Bacteria;p_Proteobacteria;c_Alphaproteobacteria; o_Sphingomonadales;f_Sphingomonadaceae; g_Sphingomonas; |
| Mycoplasma | 0.04 | 0.04 | 0.03 | 0.03 | 0.04 | 0.03 | 0.00 | 0.00 | 0.01 | k_Bacteria;p_Firmicutes;c_Bacilli;o_Mycoplasmatales; f_Mycoplasmataceae;g_Mycoplasma; |
| Escherichia-Shigella | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.02 | 0.00 | 0.04 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria;o_Enterobacterales;f_Enterobacteriaceae; g_Escherichia-Shigella; |
| Pseudomonas | 0.02 | 0.00 | 0.00 | 0.01 | 0.01 | 0.01 | 0.03 | 0.04 | 0.03 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria;o_Pseudomonadales;f_Pseudomonadaceae; g_Pseudomonas; |
| Vibrio | 0.01 | 0.00 | 0.01 | 0.03 | 0.03 | 0.04 | 0.02 | 0.03 | 0.02 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria;o_Vibrionales;f_Vibrionaceae;g_Vibrio; |
| Methylophilus | 0.03 | 0.03 | 0.03 | 0.03 | 0.02 | 0.02 | 0.00 | 0.00 | 0.00 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria;o_Burkholderiales;f_Methylophilaceae; g_Methylophilus; |
| Chloroplast | 0.00 | 0.03 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | k_Bacteria;p_Cyanobacteria;c_Cyanobacteriia; o_Chloroplast;f_Chloroplast;g_Chloroplast; |
| FukuN57 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.03 | 0.03 | k_Bacteria;p_Proteobacteria;c_Alphaproteobacteria; o_Rhizobiales;f_Beijerinckiaceae;g_FukuN57; |
| Nitrosomonas | 0.00 | 0.00 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria;o_Burkholderiales;f_Nitrosomonadaceae; g_Nitrosomonas; |
| Cupriavidus | 0.01 | 0.01 | 0.01 | 0.02 | 0.02 | 0.02 | 0.00 | 0.01 | 0.00 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria;o_Burkholderiales;f_Burkholderiaceae;g_Cupriavidus; |
| Psychrobacter | 0.016 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.02 | 0.00 | 0.00 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria;o_Pseudomonadales;f_Moraxellaceae;g_Psychrobacter; |
| Ralstonia | 0.00 | 0.00 | 0.00 | 0.02 | 0.02 | 0.02 | 0.01 | 0.01 | 0.01 | k_Bacteria;p_Proteobacteria;c_Gammaproteobacteria;o_Burkholderiales;f_Burkholderiaceae;g_Ralstonia; |
| Bacteroides | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 | 0.00 | 0.01 | k_Bacteria;p_Bacteroidota;c_Bacteroidia; o_Bacteroidales;f_Bacteroidaceae;g_Bacteroides; |
| Blautia | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | k_Bacteria;p_Firmicutes;c_Clostridia; o_Lachnospirales;f_Lachnospiraceae;g_Blautia; |
| Mitochondria | 0.00 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | k_Bacteria;p_Proteobacteria;c_Alphaproteobacteria; o_Rickettsiales;f_Mitochondria;g_Mitochondria; |
| Bifidobacterium | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | k_Bacteria;p_Actinobacteriota;c_Actinobacteria; o_Bifidobacteriales;f_Bifidobacteriaceae; g_Bifidobacterium; |
| Sample | Shannon | Simpson | Richness | Chao1 | ACE |
|---|---|---|---|---|---|
| BLX-G1.1 | 1.35 | 0.39 | 148 | 184.03 | 194.44 |
| BLX-G1.2 | 1.29 | 0.38 | 126 | 151.2 | 157.62 |
| BLX-G1.3 | 1.39 | 0.43 | 118 | 144.25 | 152.91 |
| BLX-G4.1 | 3.53 | 0.94 | 164 | 166.28 | 168.73 |
| BLX-G4.2 | 4.10 | 0.96 | 190 | 190 | 190 |
| BLX-G4.3 | 3.45 | 0.93 | 153 | 157.4 | 157.64 |
| BLX-G7.1 | 3.33 | 0.92 | 172 | 205.68 | 200.28 |
| BLX-G7.2 | 3.43 | 0.92 | 212 | 248.03 | 249.21 |
| BLX-G7.3 | 3.10 | 0.92 | 148 | 191.04 | 190.28 |
| CRP-G1.1 | 2.73 | 0.78 | 189 | 254.03 | 262.01 |
| CRP-G1.2 | 2.32 | 0.71 | 101 | 115 | 118.64 |
| CRP-G1.3 | 2.61 | 0.76 | 170 | 190.32 | 194 |
| CRP-G4.1 | 2.62 | 0.84 | 107 | 120.13 | 122.62 |
| CRP-G4.2 | 2.74 | 0.86 | 112 | 143.07 | 143.68 |
| CRP-G4.3 | 2.91 | 0.90 | 111 | 121 | 126.08 |
| CRP-G7.1 | 2.57 | 0.82 | 146 | 193.3 | 183.77 |
| CRP-G7.2 | 2.50 | 0.78 | 164 | 240 | 227.18 |
| CRP-G7.3 | 2.80 | 0.83 | 187 | 229.43 | 242.41 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, M.; Yang, W. Environmental Changes Driving Shifts in the Structure and Functional Properties of the Symbiotic Microbiota of Daphnia. Microorganisms 2024, 12, 2492. https://doi.org/10.3390/microorganisms12122492
You M, Yang W. Environmental Changes Driving Shifts in the Structure and Functional Properties of the Symbiotic Microbiota of Daphnia. Microorganisms. 2024; 12(12):2492. https://doi.org/10.3390/microorganisms12122492
Chicago/Turabian StyleYou, Minru, and Wenwu Yang. 2024. "Environmental Changes Driving Shifts in the Structure and Functional Properties of the Symbiotic Microbiota of Daphnia" Microorganisms 12, no. 12: 2492. https://doi.org/10.3390/microorganisms12122492
APA StyleYou, M., & Yang, W. (2024). Environmental Changes Driving Shifts in the Structure and Functional Properties of the Symbiotic Microbiota of Daphnia. Microorganisms, 12(12), 2492. https://doi.org/10.3390/microorganisms12122492