
Comparison of Endogenous Alpharetroviruses (ALV-like) across Galliform Species: New Distant Proviruses

Abstract
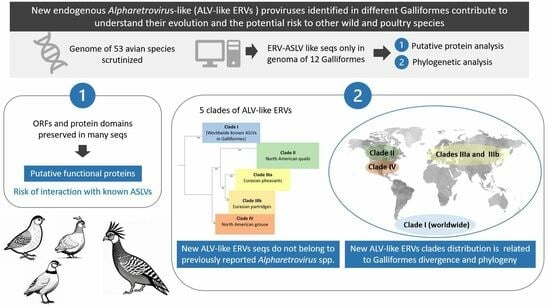
1. Introduction
2. Materials and Methods
2.1. Subject Genomes Analyzed
2.2. Data Mining
2.3. Sequence Analysis
2.4. Phylogenetic Analysis
2.5. Protein Analysis
3. Results
3.1. Genetic and Protein Analyses
3.1.1. LTR
3.1.2. gag Genes
3.1.3. pol Genes
3.1.4. env Genes
3.2. Relationships between ERVs: Phylogenetic Inference and Geographic Distribution of Their Host Species
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Payne, L.N.; Nair, V. The Long View: 40 Years of Avian Leukosis Research. Avian Pathol. 2012, 41, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Fandiño, S.; Gomez-lucia, E.; Benítez, L.; Doménech, A. Avian Leukosis: Will We Be Able to Get Rid of It? Animals 2023, 13, 2358. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Leukosis/Sarcoma Group. Diseases of Poultry; Swayne, D.E., Ed.; Wiley & Sons: Hoboken, NJ, USA, 2019; pp. 587–625. [Google Scholar]
- Gak, E.; Yaniv, A.; Sherman, L.; Ianconescu, M.; Tronick, S.R.; Gazit, A. Lymphoproliferative Disease Virus of Turkeys: Sequence Analysis and Transcriptional Activity of the Long Terminal Repeat. Gene 1991, 99, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Sarid, R.; Chajut, A.; Gak, E.; Kim, Y.; Hixson, C.V.; Oroszlan, S.; Tronick, S.R.; Gazit, A.; Abraham, Y. Genome Organization of a Biologically Active Molecular Clone of the Lymphoproliferative Disease Virus of Turkeys. Virology 1994, 204, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.B.; Kevin Keel, M.; Philips, J.E.; Cartoceti, A.N.; Munk, B.A.; Nemeth, N.M.; Welsh, T.I.; Thomas, J.M.; Crum, J.M.; Lichtenwalner, A.B.; et al. Avian Oncogenesis Induced by Lymphoproliferative Disease Virus: A Neglected or Emerging Retroviral Pathogen? Virology 2014, 450–451, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Chesters, P.M.; Howes, K.; Petherbridge, L.; Evans, S.; Payne, L.N.; Venugopal, K. The Viral Envelope Is a Major Determinant for the Induction of Lymphoid and Myeloid Tumours by Avian Leukosis Virus Subgroups A and J, Respectively. J. Gen. Virol. 2002, 83, 2553–2561. [Google Scholar] [CrossRef] [PubMed]
- Rainey, G.J.A.; Natonson, A.; Maxfield, L.F.; Coffin, J.M. Mechanisms of Avian Retroviral Host Range Extension. J. Virol. 2003, 77, 6709–6719. [Google Scholar] [CrossRef]
- Weber, F.; Schaffner, W. Enhancer Activity Correlates with the Oncogenic Potential of Avian Retroviruses. EMBO J. 1985, 4, 949–956. [Google Scholar] [CrossRef]
- Malik, H.S.; Henikoff, S.; Eickbush, T.H. Poised for Contagion: Evolutionary Origins of the Infectious Abilities of Invertebrate Retroviruses. Genome Res. 2000, 10, 1307–1318. [Google Scholar] [CrossRef]
- Gifford, R.J.; Blomberg, J.; Coffin, J.M.; Fan, H.; Heidmann, T.; Mayer, J.; Stoye, J.; Tristem, M.; Johnson, W.E. Nomenclature for Endogenous Retrovirus (ERV) Loci. Retrovirology 2018, 15, 59. [Google Scholar] [CrossRef]
- Mason, A.S.; Lund, A.R.; Hocking, P.M.; Fulton, J.E.; Burt, D.W.; Burt, D.W. Identification and Characterisation of Endogenous Avian Leukosis Virus Subgroup E (ALVE) Insertions in Chicken Whole Genome Sequencing Data. Mob. DNA 2020, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.N. Biology of Avian Retroviruses. In The Retroviridae; Levy, J.A., Ed.; Plenum Press: New York, NY, USA, 1992; Volume 1, pp. 299–404. [Google Scholar]
- Frossard, J.P. Retroviridae, 4th ed.; McVey, D.S., Kennedy, M., Chengappa, M.M., Wilkes, R., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2022; ISBN 9781119650751. [Google Scholar]
- Hu, X.; Zhu, W.; Chen, S.; Liu, Y.; Sun, Z.; Gao, B. Expression Patterns of Endogenous Avian Retrovirus ALVE1 and Its Response to Infection with Exogenous Avian Tumour Viruses. Arch. Virol. 2016, 162, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Hanafusa, T.; Hanafusa, H. Isolation of Leukosis-Type Virus from Pheasant Embryo Cells: Possible Presence of Viral Genes in Cells. Virology 1973, 51, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Fujita, D.J.; Chen, Y.C.; Friis, R.R.; Vogt, P.K. RNA Tumor Viruses of Pheasants: Characterization of Avian Leukosis Subgroups F and G. Virology 1974, 60, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Hanafusa, T.; Hanafusa, H.; Metroka, C.E.; Hayward, W.S.; Rettenmier, C.W.; Sawyer, R.C.; Dougherty, R.M.; Distefano, H.S. Pheasant Virus: New Class of Ribodeoxyvirus. Proc. Natl. Acad. Sci. USA 1976, 73, 1333–1337. [Google Scholar] [CrossRef]
- Troesch, C.D.; Vogt, P.K. An Endogenous Virus from Lophortyx Quail Is the Prototype for Envelope Subgroup I of Avian Retroviruses. Virology 1985, 143, 595–602. [Google Scholar] [CrossRef]
- Chen, Y.C.; Vogt, P.K. Endogenous Leukosis Viruses in the Avian Family Phasianidae. Virology 1977, 76, 740–750. [Google Scholar] [CrossRef]
- Dimcheff, D.E.; Drovetski, S.V.; Krishnan, M.; Mindell, D.P. Cospeciation and Horizontal Transmission of Avian Sarcoma and Leukosis Virus Gag Genes in Galliform Birds. J. Virol. 2000, 74, 3984–3995. [Google Scholar] [CrossRef]
- Dimcheff, D.E.; Krishnan, M.; Mindell, D.P. Evolution and Characterization of Tetraonine Endogenous Retrovirus: A New Virus Related to Avian Sarcoma and Leukosis Viruses. J. Virol. 2001, 75, 2002–2009. [Google Scholar] [CrossRef]
- Mays, J.K.; Black-Pyrkosz, A.; Mansour, T.; Schutte, B.C.; Chang, S.; Dong, K.; Hunt, H.D.; Fadly, A.M.; Zhang, L.; Zhang, H. Endogenous Avian Leukosis Virus in Combination with Serotype 2 Marek’ s Disease Virus Significantly Boosted the Incidence of Lymphoid Leukosis-Like Bursal Lymphomas in Susceptible Chickens. J. Virol. 2019, 93, e00861-19. [Google Scholar] [CrossRef]
- Ramoutar, V.V.; Johnson, Y.J.; Kohrt, L.J.; Bahr, J.M.; Caporali, E.H.G.; Myint, M.S.; Szigetvari, N.; Matthew, C.; Ramoutar, V.V.; Johnson, Y.J.; et al. Retroviral Association with Ovarian Adenocarcinoma in Laying Hens. Avian Pathol. 2022, 51, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Fulton, J.E.; Mason, A.S.; Wolc, A.; Arango, J.; Settar, P.; Lund, A.R.; Burt, D.W. The Impact of Endogenous Avian Leukosis Viruses (ALVE) on Production Traits in Elite Layer Lines. Poult. Sci. 2021, 100, 101121. [Google Scholar] [CrossRef] [PubMed]
- Gavora, J.S.; Kuhnlein, U.; Crittenden, L.B.; Spencer, J.L.; Sabour, M.P. Endogenous Viral Genes: Association with Reduced Egg Production Rate 20 Loci Have Been Distinguished in White Leghorns as DNA Restriction Fragment Length Proviral DNA Sequences, Residing Perma-Polymorphisms and Many More Have Been Nently in the Individ. Poult. Sci. 1991, 70, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Chiu, E.S.; Vandewoude, S. Endogenous Retroviruses Drive Resistance and Promotion of Exogenous Retroviral Homologs. Annu. Rev. Anim. Biosci. 2021, 9, 225–248. [Google Scholar] [CrossRef]
- Aswad, A.; Katzourakis, A. Paleovirology and Virally Derived Immunity. Trends Ecol. Evol. 2012, 27, 627–636. [Google Scholar] [CrossRef]
- Mason, A.S.; Miedzinska, K.; Kebede, A.; Bamidele, O.; Al-Jumaili, A.S.; Dessie, T.; Hanotte, O.; Smith, J. Diversity of Endogenous Avian Leukosis Virus Subgroup e (ALVE) Insertions in Indigenous Chickens. Genet. Sel. Evol. 2020, 52, 29. [Google Scholar] [CrossRef]
- Payne, L.N.; Brown, S.R.; Bumstead, N.; Howes, K.; Frazier, J.A.; Thouless, M.E. A Novel Subgroup of Exogenous Avian Leukosis Virus in Chickens. J. Gen. Virol. 1991, 72, 801–807. [Google Scholar] [CrossRef]
- Wang, X.; Peng, Z.; Zhi-Zhong, C. Identification of a New Subgroup of Avian Leukosis Virus Isolated from Chinese Indigenous Chicken Breeds. Chinese J. Virol. 2012, 28, 609–614. [Google Scholar]
- Bai, J.; Payne, L.N.; Skinner, M.A. HPRS-103 (Exogenous Avian Leukosis Virus, Subgroup J) Has an Env Gene Related to Those of Endogenous Elements EAV-0 and E51 and an E Element Found Previously Only in Sarcoma Viruses. J. Virol. 1995, 69, 779–784. [Google Scholar] [CrossRef]
- Cai, L.; Shen, Y.; Wang, G.; Guo, H.; Liu, J.; Cheng, Z. Identification of Two Novel Multiple Recombinant Avian Leukosis Viruses in Two Different Lines of Layer Chicken. J. Gen. Virol. 2013, 94, 2278–2286. [Google Scholar] [CrossRef][Green Version]
- Wang, P.; Niu, J.; Xue, C.; Han, Z.; Abdelazez, A.; Xinglin, Z. Two Novel Recombinant Avian Leukosis Virus Isolates from Luxi Gamecock Chickens. Arch. Virol. 2020, 165, 2877–2881. [Google Scholar] [CrossRef] [PubMed]
- Gould, W.J.; O’connell, P.H.; Shivaprasad, H.L.; Schat, K.A. Detection of Retrovirus Sequences in Budgerigars with Tumours. Avian Pathol. 1993, 22, 33–45. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, R.A.; Pérez-Martínez, C.; Espinosa-Alvarez, J.; Escudero-Diez, A.; García-Marín, J.F.; Núñez, A.; García-Iglesias, M.J. Lymphoid Leukosis in an Ostrich (Struthio Camelus). Vet. Rec. 2000, 146, 676–677. [Google Scholar] [CrossRef] [PubMed]
- Vařejka, F.; Tomšik, F. The Role of House Sparrow (Passer Domesticus L.) in the Spread of Leukosis Viruses in Poultry. I. Determination of Neutralizing Antibodies. ACTA Vet. BRNO 1974, 43, 367–370. [Google Scholar]
- Jiang, L.; Zeng, X.; Hua, Y.; Gao, Q.; Fan, Z.; Chai, H.; Wang, Q.; Qi, X.; Wang, Y.; Gao, H.; et al. Genetic Diversity and Phylogenetic Analysis of Glycoprotein Gp85 of Avian Leukosis Virus Subgroup J Wild-Bird Isolates from Northeast China. Arch. Virol. 2014, 159, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Hao, R.; Han, C.; Liu, L.; Zeng, X. First Finding of Subgroup-E Avian Leukosis Virus from Wild Ducks in China. Vet. Microbiol. 2014, 173, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Sperber, G.O.; Airola, T.; Jern, P.; Blomberg, J. Automated Recognition of Retroviral Sequences in Genomic Data-RetroTector©. Nucleic Acids Res. 2007, 35, 4964–4976. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, H.; Gong, Z.; Han, G.Z. Endogenous Retroviruses of Non-Avian/Mammalian Vertebrates Illuminate Diversity and Deep History of Retroviruses. PLoS Pathog. 2018, 14, e1007072. [Google Scholar] [CrossRef]
- Guoqing, L.; Moriyama, E.N. Software Review Vector NTI, a Balanced All-in-One Sequence Analysis Suite. Brief. Bioinform. 2004, 5, 378–388. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Brunak, S.; Engelbrecht, J.; Knudsen, S. Prediction of Human MRNA Donor and Acceptor Sites from the DNA Sequence. J. Mol. Biol. 1991, 220, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Yang, Z.; Li, H.; Hong, Y.; Fan, D.; Lin, A.; Xiang, L.; Shao, J. Genome-Wide Characterization of Zebrafish Endogenous Retroviruses Reveals Unexpected Diversity in Genetic Organizations and Functional Potentials. Microbiol. Spectr. 2021, 9, e0225421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative Genomics Reveals Insights into Avian Genome Evolution and Adaptation. Science 2014, 346, 1311–1321. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The Conserved Domain Database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Belshaw, R.; Watson, J.; Katzourakis, A.; Howe, A.; Woolven-Allen, J.; Burt, A.; Tristem, M. Rate of Recombinational Deletion among Human Endogenous Retroviruses. J. Virol. 2007, 81, 9437–9442. [Google Scholar] [CrossRef]
- Hogan, V.; Johnson, W.E. Ubiquitous Gamma-Type Envelope Glycoprotein. Viruses. 2023, 15, 274. [Google Scholar] [CrossRef]
- Baratti, M.; Ammannati, M.; Magnelli, C.; Dessì-Fulgheri, F. Introgression of Chukar Genes into a Reintroduced Red-Legged Partridge (Alectoris Rufa) Population in Central Italy. Anim. Genet. 2005, 36, 29–35. [Google Scholar] [CrossRef]
- Stoye, J.P. Studies of Endogenous Retroviruses Reveal a Continuing Evolutionary Saga. Nat. Rev. Microbiol. 2012, 10, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Kauff, F.; Gabaldón, T. A Phylogenomics Approach for Selecting Robust Sets of Phylogenetic Markers. Nucleic Acids Res. 2014, 42, e54. [Google Scholar] [CrossRef] [PubMed]
- Benachenhou, F.; Sperber, G.O.; Bongcam-Rudloff, E.; Andersson, G.; Boeke, J.D.; Blomberg, J. Conserved Structure and Inferred Evolutionary History of Long Terminal Repeats (LTRs). Mob. DNA 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.; McElliott, V.; Vapniarsky, N.; Oliver, A.; Rowe, J. Tissue Tropism and Promoter Sequence Variation in Caprine Arthritis Encephalitis Virus Infected Goats. Virus Res. 2010, 151, 177–184. [Google Scholar] [CrossRef]
- Balvay, L.; Lastra, M.L.; Sargueil, B.; Darlix, J.L.; Ohlmann, T. Translational Control of Retroviruses. Nat. Rev. Microbiol. 2007, 5, 128–140. [Google Scholar] [CrossRef]
- Coffin, J.; Blomberg, J.; Fan, H.; Gifford, R.; Hatziioannou, T.; Lindemann, D.; Mayer, J.; Stoye, J.; Tristem, M.; Johnson, W. ICTV Virus Taxonomy Profile: Retroviridae 2021. J. Gen. Virol. 2021, 102, 001712. [Google Scholar] [CrossRef]
- Wang, N.; Kimball, R.T.; Braun, E.L.; Liang, B.; Zhang, Z. Assessing Phylogenetic Relationships among Galliformes: A Multigene Phylogeny with Expanded Taxon Sampling in Phasianidae. PLoS ONE 2013, 8, e64312. [Google Scholar] [CrossRef]
- Chen, D.; Hosner, P.A.; Dittmann, D.L.; O’Neill, J.P.; Birks, S.M.; Braun, E.L.; Kimball, R.T. Divergence Time Estimation of Galliformes Based on the Best Gene Shopping Scheme of Ultraconserved Elements. BMC Ecol. Evol. 2021, 21, 209. [Google Scholar] [CrossRef]
- Chen, S.; Wu, T.; Wang, D.; Wang, B. An Endogenous Retroviral LTR-Derived Long Noncoding RNA Lnc-LTR5B Interacts with BiP to Modulate ALV-J Replication in Chicken Cells. Front. Microbiol. 2021, 12, 788317. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Blair Hedges, S. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Li, X.; Huang, Y.; Lei, F. Comparative Mitochondrial Genomics and Phylogenetic Relationships of the Crossoptilon Species (Phasianidae, Galliformes). BMC Genomics 2015, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.W.; Brown, J.W.; Mooers, A.O. A Molecular Genetic Time Scale Demonstrates Cretaceous Origins and Multiple Diversification Rate Shifts within the Order Galliformes (Aves). Mol. Phylogenet. Evol. 2015, 92, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A. Origin of the Retroviruses: When, Where, and How? Curr. Opin. Virol. 2017, 25, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Katzourakis, A.; Rambaut, A.; Pybus, O.G. The Evolutionary Dynamics of Endogenous Retroviruses. Trends Microbiol. 2005, 13, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Fujita, D.J.; Tal, J.; Varmus, H.E.; Bishop, J.M. Env Gene of Chicken RNA Tumor Viruses: Extent of Conservation in Cellular and Viral Genomes. J. Virol. 1978, 27, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Postler, T.S.; Desrosiers, R.C. The Tale of the Long Tail: The Cytoplasmic Domain of HIV-1 Gp41. J. Virol. 2013, 87, 2–15. [Google Scholar] [CrossRef]
- Tedbury, P.R.; Freed, E.O. The Cytoplasmic Tail of Retroviral Envelope Glycoproteins. Prog. Mol. Biol. Transl. Sci. 2015, 129, 253–284. [Google Scholar] [CrossRef]
- Salter, J.F.; Hosner, P.A.; Tsai, W.L.E.; McCormack, J.E.; Braun, E.L.; Kimball, R.T.; Brumfield, R.T.; Faircloth, B.C. Historical Specimens and the Limits of Subspecies Phylogenomics in the New World Quails (Odontophoridae). Mol. Phylogenet. Evol. 2022, 175, 107559. [Google Scholar] [CrossRef]
- Hosner, P.A.; Braun, E.L.; Kimball, R.T. Land Connectivity Changes and Global Cooling Shaped the Colonization History and Diversification of New World Quail (Aves: Galliformes: Odontophoridae). J. Biogeogr. 2015, 42, 1883–1895. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Subfamily | Scientific Name | Species | ERV Structure | Potentially Functional ORFs | ERV Size (Kbp) | Number of Sequences |
|---|---|---|---|---|---|---|---|
| Odontophoridae | Callipepla californica | California quail | LTR5′-gag-pol-env-LTR3′ | gag, pol, env | 7.6 | 5 | |
| Odontophoridae | Callipepla squamata | Scaled quail | LTR5′-gag-pol | gag, pol | 5.4 1 | 4 | |
| Odontophoridae | Colinus virginianus | Virginia quail | ?-LTR3′ 2 | Unknown | 1 | ||
| Phasianidae | Perdicinae | Alectoris rufa | Red-legged partridge | LTR5′-gag-pol-env-LTR3′ | gag, pol, env | 7.4 | 2 |
| Phasianidae | Perdicinae | Alectoris magna | Rusty-necklaced partridge | LTR5′-gag-pol-env-LTR3′ | gag, pol, env | 7.4 | 4 |
| Phasianidae | Tetraoninae | Centrocercus urophasianus | Greater sage-grouse | LTR5′-gag-pol-env-LTR3′ 3 | pol, env | 7.7 1 | 2 |
| Phasianidae | Tetraoninae | Centrocercus minimus | Lesser sage-grouse | LTR5′-gag-pol-env-LTR3′ 3 | 7.6 1 | 2 | |
| Phasianidae | Tetraoninae | Lagopus leucura | White-tailed ptarmigan | LTR5′-gag-env-LTR3′ | 4.8 | 3 | |
| Phasianidae | Tetraoninae | Lagopus muta | Rock ptarmigan | LTR5′-gag-env-LTR3′ | env | 5.1 | 2 |
| Phasianidae | Phasianinae | Phasianus colchicus | Ring-necked pheasant | LTR5′-gag-pol-env-LTR3′ | gag, pol, env | 7.5 | 6 |
| Phasianidae | Tetraoninae | Tympanuchus pallidicinctus | Lesser prairie chicken | LTR5′-gag-env-LTR3′ | 3.9 | 1 | |
| Phasianidae | Tetraoninae | Tympanuchus cupido | Greater prairie chicken | LTR5′-gag-env 4 | 3.2 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fandiño, S.; Gomez-Lucia, E.; Benítez, L.; Doménech, A. Comparison of Endogenous Alpharetroviruses (ALV-like) across Galliform Species: New Distant Proviruses. Microorganisms 2024, 12, 86. https://doi.org/10.3390/microorganisms12010086
Fandiño S, Gomez-Lucia E, Benítez L, Doménech A. Comparison of Endogenous Alpharetroviruses (ALV-like) across Galliform Species: New Distant Proviruses. Microorganisms. 2024; 12(1):86. https://doi.org/10.3390/microorganisms12010086
Chicago/Turabian StyleFandiño, Sergio, Esperanza Gomez-Lucia, Laura Benítez, and Ana Doménech. 2024. "Comparison of Endogenous Alpharetroviruses (ALV-like) across Galliform Species: New Distant Proviruses" Microorganisms 12, no. 1: 86. https://doi.org/10.3390/microorganisms12010086
APA StyleFandiño, S., Gomez-Lucia, E., Benítez, L., & Doménech, A. (2024). Comparison of Endogenous Alpharetroviruses (ALV-like) across Galliform Species: New Distant Proviruses. Microorganisms, 12(1), 86. https://doi.org/10.3390/microorganisms12010086