

Distribution and Implications of Haloarchaeal Plasmids Disseminated in Self-Encoded Plasmid Vesicles

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling of Australian Salt Lakes, DNA Extraction, and Sequencing
2.2. Creation of pR1SE Protein Clusters
2.3. Detection of pR1SE-like Elements (apHPVs) in Public Databases and Metagenomes from Australian Salt Lakes
2.4. Identification of the apHPV Core Genes
2.5. Phylogenetic Analysis
2.6. Gene Sharing Network of apHPVs, Archaeal Viruses, and Archaeal Plasmids
2.7. Functional Annotation of apHPV Genes
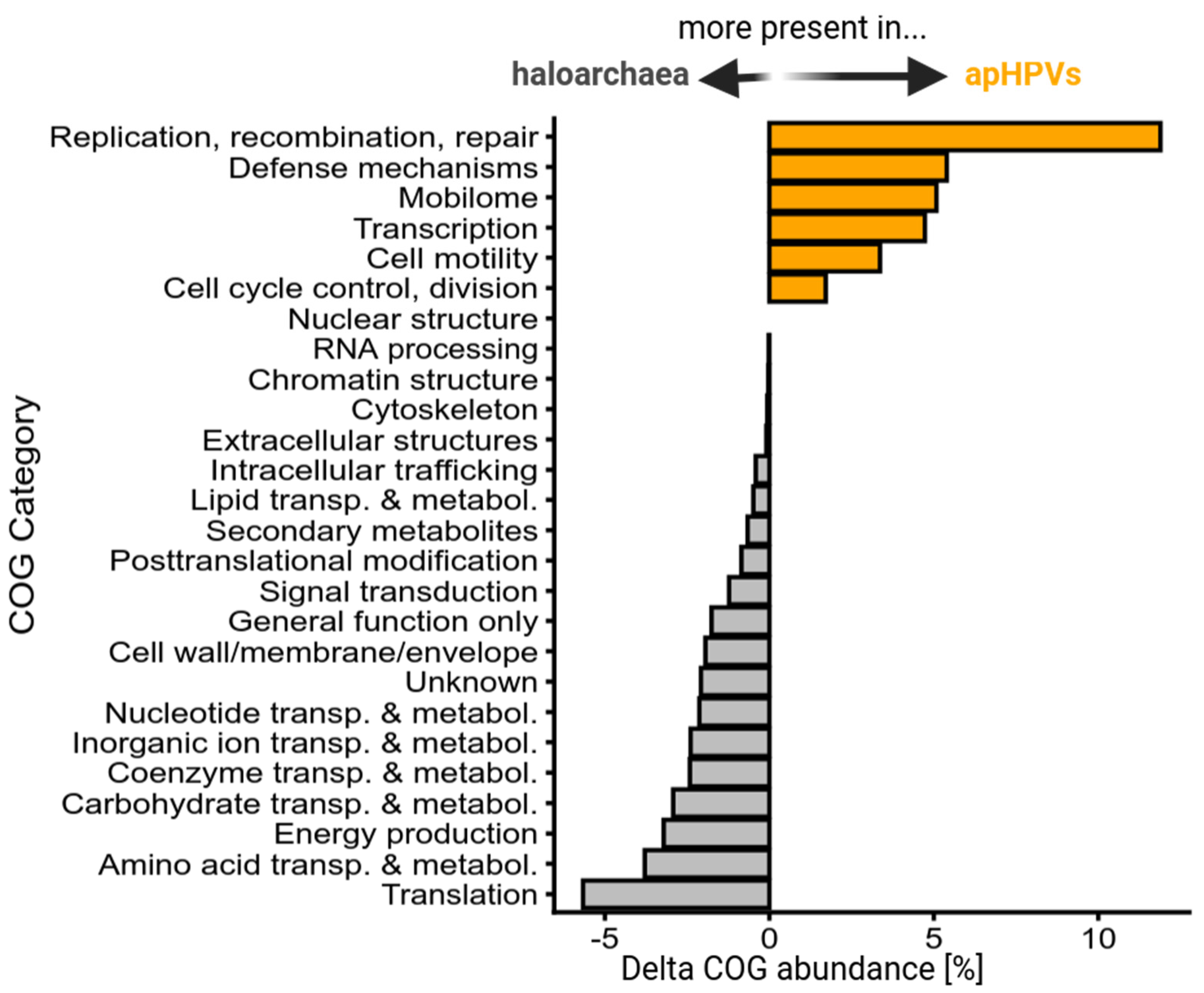
2.8. Identification of Clusters of Orthologous Groups (COGs)
2.9. Detection of Antiviral Defense Systems and Anti-CRISPR Proteins in apHPVs
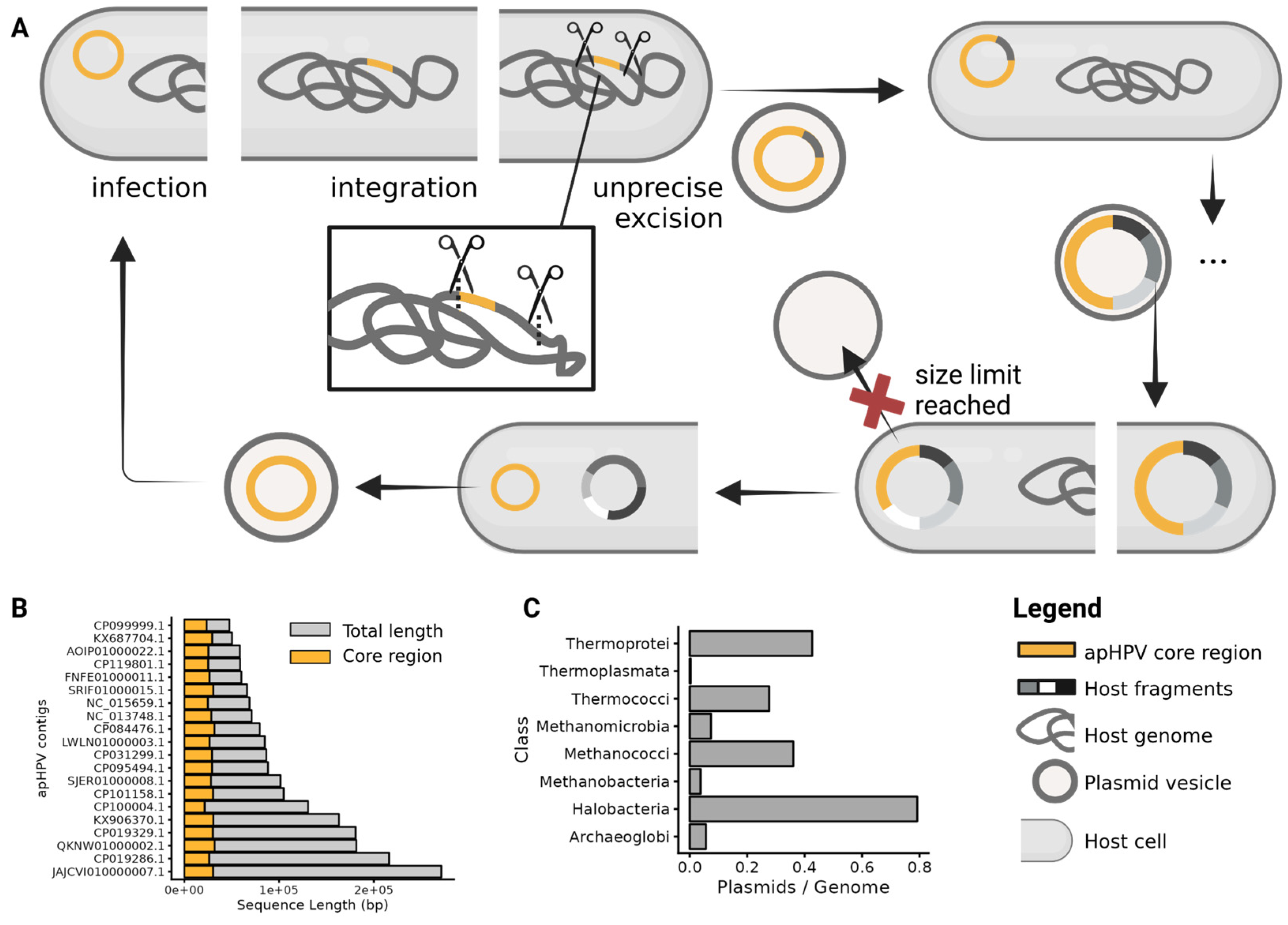
2.10. Number of Plasmids per Class
3. Results
3.1. Selection of Core Proteins of pR1SE
3.2. Identification of Forty Complete pR1SE-like Mobile Genetic Elements Using an HMM-Based Approach
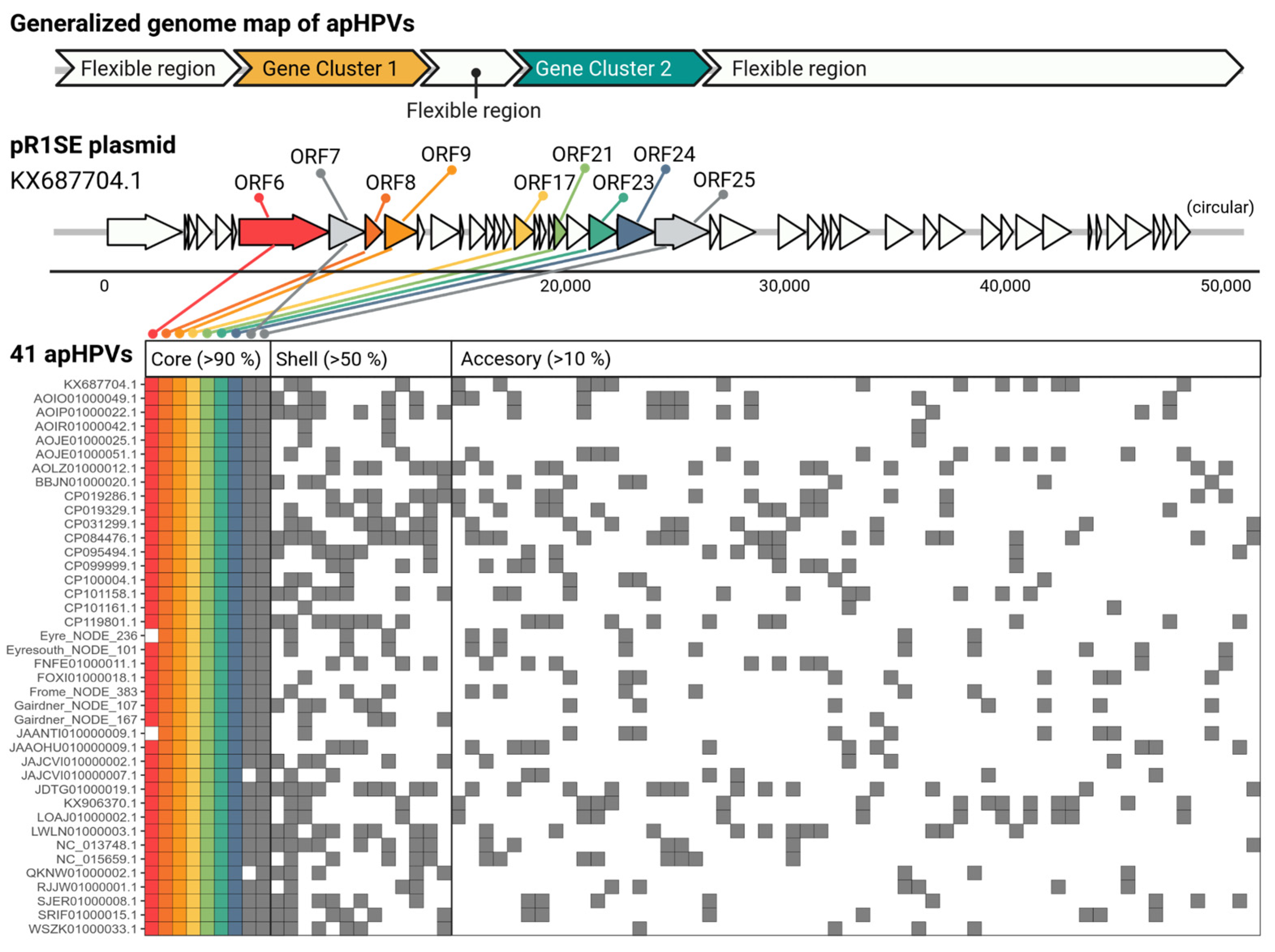
3.3. apHPVs Exhibit Two Highly Conserved Gene Clusters Interspersed by a Variable Region
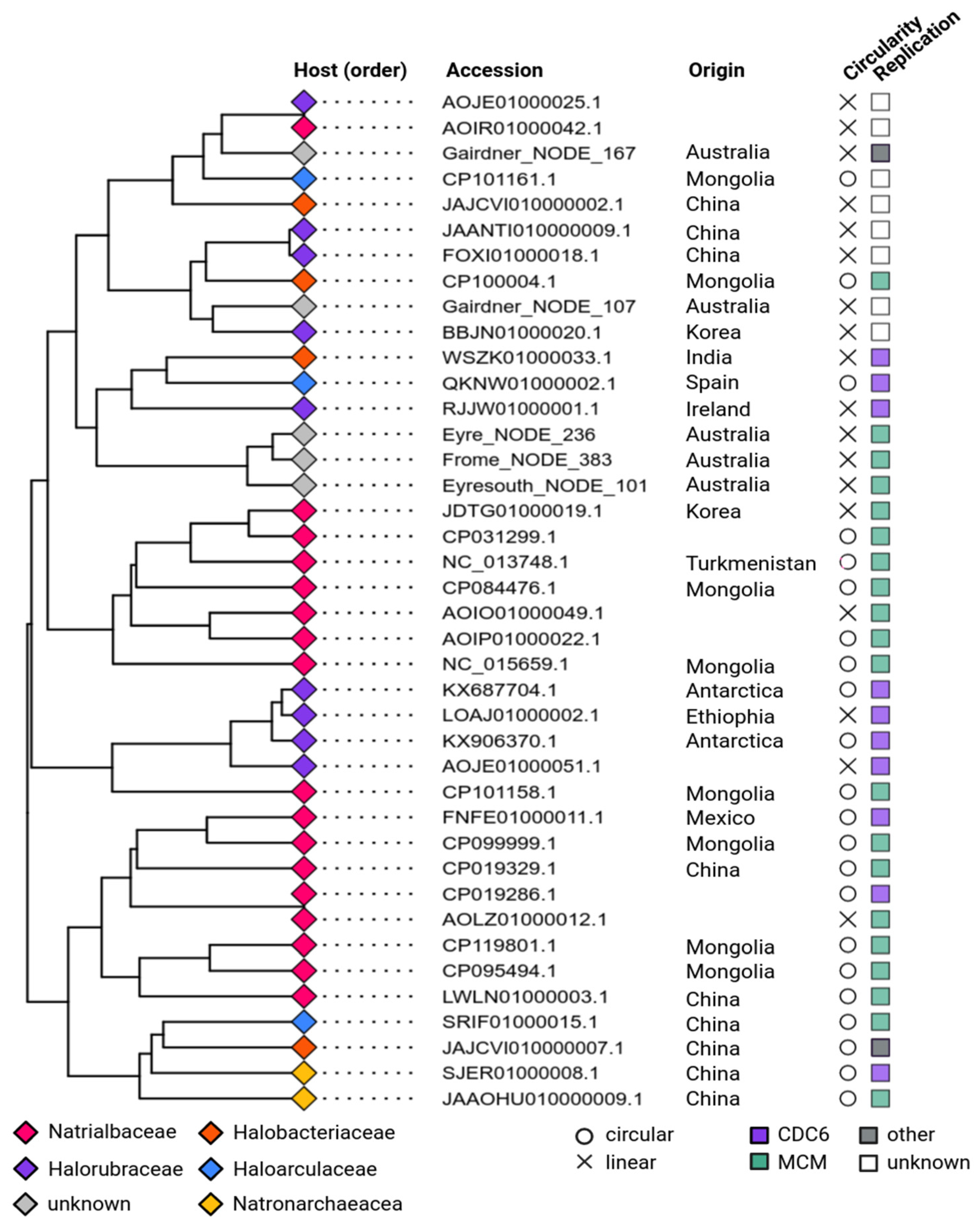
3.4. Phylogeny and Host Association of apHPVs
3.5. Replication of apHPVs
3.6. Core Genes and Their Predicted Functions
3.7. Transfer of Host Genetic Material by apHPVs
3.8. Antiviral Defense Systems and Anti-CRISPR Proteins on apHPVs
3.9. Clustering of apHPVs with Archaeal Plasmids and Archaeal Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hall, C.; Brachat, S.; Dietrich, F.S. Contribution of Horizontal Gene Transfer to the Evolution of Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 1102–1115. [Google Scholar] [CrossRef] [PubMed]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Kuhn, J.H. Viruses Defined by the Position of the Virosphere within the Replicator Space. Microbiol. Mol. Biol. Rev. 2021, 85, e00193-20. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, S.; Tschitschko, B.; Zhong, L.; Raftery, M.J.; Cavicchioli, R. A plasmid from an Antarctic haloarchaeon uses specialized membrane vesicles to disseminate and infect plasmid-free cells. Nat. Microbiol. 2017, 2, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.; Gebhard, L.J.; Schubotz, F.; Shevchenko, A.; Speth, D.R.; Liao, Y.; Duggin, I.G.; Marchfelder, A.; Erdmann, S. Extracellular vesicles of Euryarchaeida: Precursor to eukaryotic membrane trafficking. bioRxiv 2023. [Google Scholar] [CrossRef]
- Gaudin, M.; Gauliard, E.; Schouten, S.; Houel-Renault, L.; Lenormand, P.; Marguet, E.; Forterre, P. Hyperthermophilic archaea produce membrane vesicles that can transfer DNA. Environ. Microbiol. Rep. 2013, 5, 109–116. [Google Scholar] [CrossRef]
- Gebhard, L.J.; Vershinin, Z.; Alarcón-Schumacher, T.; Eichler, J.; Erdmann, S. Influence of N-Glycosylation on Virus–Host Interactions in Halorubrum lacusprofundi. Viruses 2023, 15, 1469. [Google Scholar] [CrossRef] [PubMed]
- DeMaere, M.Z.; Williams, T.J.; Allen, M.A.; Brown, M.V.; Gibson, J.A.E.; Rich, J.; Lauro, F.M.; Dyall-Smith, M.; Davenport, K.W.; Woyke, T.; et al. High level of intergenera gene exchange shapes the evolution of haloarchaea in an isolated Antarctic lake. Proc. Natl. Acad. Sci. USA 2013, 110, 16939–16944. [Google Scholar] [CrossRef]
- Forterre, P.; Krupovic, M.; Raymann, K.; Soler, N. Plasmids from Euryarchaeota. Microbiol. Spectr. 2014, 2, 27. [Google Scholar] [CrossRef]
- Alarcón-Schumacher, T.; Lücking, D.; Erdmann, S. Revisiting evolutionary trajectories and the organization of the Pleolipoviridae family. PLoS Genet. 2023, 19, e1010998. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Kazutaka, K.; Misakwa, K.; Kei-ichi, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Camargo, A.P.; Nayfach, S.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Ritter, S.J.; Reddy, T.B.K.; Mukherjee, S.; Schulz, F.; et al. IMG/VR v4: An expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic Acids Res. 2023, 51, D733–D743. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Camargo, A.P.; Coutinho, F.H.; Dabdoub, S.M.; Dutilh, B.E.; Nayfach, S.; Tritt, A. iPHoP: An integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria. PLoS Biol. 2023, 21, e3002083. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Yu, G. Data Integration, Manipulation and Visualization of Phylogenetic Trees; CRC Press: Boca Raton, FL, USA, 2022. [Google Scholar]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory friendly classification with the genome taxonomy database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Guo, J.; Vik, D.; Pratama, A.A.; Roux, S.; Sullivan, M. Viral Sequence Identification SOP with VirSorter2. July 2021. Available online: https://www.protocols.io/view/viral-sequence-identification-sop-with-virsorter2-bwm5pc86 (accessed on 21 March 2023).
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, M.; Borton, M.A.; McGivern, B.B.; Zayed, A.A.; La Rosa, S.L.; Solden, L.M.; Liu, P.; Narrowe, A.B.; Rodríguez-Ramos, J.; Bolduc, B.; et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- van Kempen, M.; Kim, S.S.; Tumescheit, C.; Mirdita, M.; Lee, J.; Gilchrist, C.L.M.; Söding, J.; Steinegger, M. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 2023, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Laiho, A.; Törönen, P.; Salgado, M. DALI shines a light on remote homologs: One hundred discoveries. Protein Sci. 2023, 32, e4519. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Alvarez, R.V.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2020, 49, D274–D281. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.J.; Meaden, S.; Mestre, M.R.; Palmer, C.; Toro, N.; Fineran, P.C.; A Jackson, S. PADLOC: A web server for the identification of antiviral defence systems in microbial genomes. Nucleic Acids Res. 2022, 50, W541–W550. [Google Scholar] [CrossRef]
- Dong, C.; Wang, X.; Ma, C.; Zeng, Z.; Pu, D.-K.; Liu, S.; Wu, C.-S.; Chen, S.; Deng, Z.; Guo, F.-B. Anti-CRISPRdb v2.2: An online repository of anti-CRISPR proteins including information on inhibitory mechanisms, activities and neighbors of curated anti-CRISPR proteins. Database 2022, 2022, baac010. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, H.; Liu, J.; Liu, X.; Xiang, H. Diversity and evolution of multiple orc/cdc6-adjacent replication origins in haloarchaea. BMC Genom. 2012, 13, 478. [Google Scholar] [CrossRef] [PubMed]
- Rzechorzek, N.J.; Blackwood, J.K.; Bray, S.M.; Maman, J.D.; Pellegrini, L.; Robinson, N.P. Structure of the hexameric HerA ATPase reveals a mechanism of translocation-coupled DNA-end processing in archaea. Nat. Commun. 2014, 5, 5506. [Google Scholar] [CrossRef] [PubMed]
- Happonen, L.J.; Oksanen, E.; Liljeroos, L.; Goldman, A.; Kajander, T.; Butcher, S.J. The Structure of the NTPase That Powers DNA Packaging into Sulfolobus Turreted Icosahedral Virus 2. J. Virol. 2013, 87, 8388–8398. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR–Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Melamed, S.; Millman, A.; Shulman, G.; Oppenheimer-Shaanan, Y.; Kacen, A.; Doron, S.; Amitai, G.; Sorek, R. Cyclic GMP–AMP signalling protects bacteria against viral infection. Nature 2019, 574, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Millman, A.; Melamed, S.; Amitai, G.; Sorek, R. Diversity and classification of cyclic-oligonucleotide-based anti-phage signalling systems. Nat. Microbiol. 2020, 5, 1608–1615. [Google Scholar] [CrossRef]
- Wang, L.; Chen, S.; Xu, T.; Taghizadeh, K.; Wishnok, J.S.; Zhou, X.; Deng, Z.; Dedon, P.C. Phosphorothioation of DNA in bacteria by dnd genes. Nat. Chem. Biol. 2007, 3, 709–710. [Google Scholar] [CrossRef]
- Xu, T.; Yao, F.; Zhou, X.; Deng, Z.; You, D. A novel host-specific restriction system associated with DNA backbone S-modification in Salmonella. Nucleic Acids Res. 2010, 38, 7133–7141. [Google Scholar] [CrossRef]
- Millman, A.; Melamed, S.; Leavitt, A.; Doron, S.; Bernheim, A.; Hör, J.; Garb, J.; Bechon, N.; Brandis, A.; Lopatina, A.; et al. An expanded arsenal of immune systems that protect bacteria from phages. Cell Host Microbe 2022, 30, 1556–1569.e5. [Google Scholar] [CrossRef]
- Lima-Mendez, G.; Toussaint, A.; Leplae, R. A modular view of the bacteriophage genomic space: Identification of host and lifestyle marker modules. Res. Microbiol. 2011, 162, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M. Paradigms of plasmid organization. Mol. Microbiol. 2000, 37, 485–491. [Google Scholar] [CrossRef]
- Mercier, C.; Thies, D.; Zhong, L.; Raftery, M.J.; Erdmann, S. Characterization of an archaeal virus-host system reveals massive genomic rearrangements in a laboratory strain. Front. Microbiol. 2023, 14, 1274068. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, M.; Malla, S.; Blythe, M.J.; Nieduszynski, C.A.; Allers, T. Accelerated growth in the absence of DNA replication origins. Nature 2013, 503, 544–547. [Google Scholar] [CrossRef]
- Eugster, A.; Frigerio, G.; Dale, M.; Duden, R. The α- and β′-COP WD40 Domains Mediate Cargo-selective Interactions with Distinct Di-lysine Motifs. Mol. Biol. Cell 2004, 15, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Shen, N.; Pei, J.; Grishin, N.V. Double-stranded DNA bacteriophage prohead protease is homologous to herpesvirus protease. Protein Sci. 2004, 13, 2260–2269. [Google Scholar] [CrossRef]
- Ottmann, C.; Luberacki, B.; Küfner, I.; Koch, W.; Brunner, F.; Weyand, M.; Mattinen, L.; Pirhonen, M.; Anderluh, G.; Seitz, H.U.; et al. A common toxin fold mediates microbial attack and plant defense. Proc. Natl. Acad. Sci. USA 2009, 106, 10359–10364. [Google Scholar] [CrossRef]
- Rutter, B.D.; Innes, R.W. Extracellular vesicles in phytopathogenic fungi. Extracell. Vesicles Circ. Nucleic Acids 2023, 4, 90–106. [Google Scholar] [CrossRef]
- Tschitschko, B.; Erdmann, S.; DeMaere, M.Z.; Roux, S.; Panwar, P.; Allen, M.A.; Williams, T.J.; Brazendale, S.; Hancock, A.M.; Eloe-Fadrosh, E.A.; et al. Genomic variation and biogeography of Antarctic haloarchaea. Microbiome 2018, 6, 113. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORF No. 1 | Pairwise AAI 2 | Best Hit | Method of Annotation |
|---|---|---|---|
| ORF6 | 18.3% | - | AlphaFold/DALI |
| ORF7 | 29.1% | subtilisin-like serine protease | foldseek/AlphaFold/DALI |
| ORF8 | 23.8% | Necrosis and ethylene inducing protein 1 | foldseek |
| ORF9 | 17.5% | S-layer/Big6/CotH | foldseek |
| ORF17 | 34.2% | PKD/SH3/Ricin B-type lectin/UnbV_ASPIC | foldseek |
| ORF21 | 26% | - | foldseek |
| ORF23 | 31.5% | pilus assembly proteins/TonB receptor proteins | foldseek |
| ORF24 | 23.9% | - | foldseek |
| ORF25 | 41% | VirB4/helicase/genome packaging ATPase | foldseek/AlphaFold/DALI |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lücking, D.; Alarcón-Schumacher, T.; Erdmann, S. Distribution and Implications of Haloarchaeal Plasmids Disseminated in Self-Encoded Plasmid Vesicles. Microorganisms 2024, 12, 5. https://doi.org/10.3390/microorganisms12010005
Lücking D, Alarcón-Schumacher T, Erdmann S. Distribution and Implications of Haloarchaeal Plasmids Disseminated in Self-Encoded Plasmid Vesicles. Microorganisms. 2024; 12(1):5. https://doi.org/10.3390/microorganisms12010005
Chicago/Turabian StyleLücking, Dominik, Tomás Alarcón-Schumacher, and Susanne Erdmann. 2024. "Distribution and Implications of Haloarchaeal Plasmids Disseminated in Self-Encoded Plasmid Vesicles" Microorganisms 12, no. 1: 5. https://doi.org/10.3390/microorganisms12010005
APA StyleLücking, D., Alarcón-Schumacher, T., & Erdmann, S. (2024). Distribution and Implications of Haloarchaeal Plasmids Disseminated in Self-Encoded Plasmid Vesicles. Microorganisms, 12(1), 5. https://doi.org/10.3390/microorganisms12010005