
Fused Enzyme Glucose-6-Phosphate Dehydrogenase::6-Phosphogluconolactonase (G6PD::6PGL) as a Potential Drug Target in Giardia lamblia, Trichomonas vaginalis, and Plasmodium falciparum

 , , , ,
, , , ,  ,
,  , and
, and
Abstract
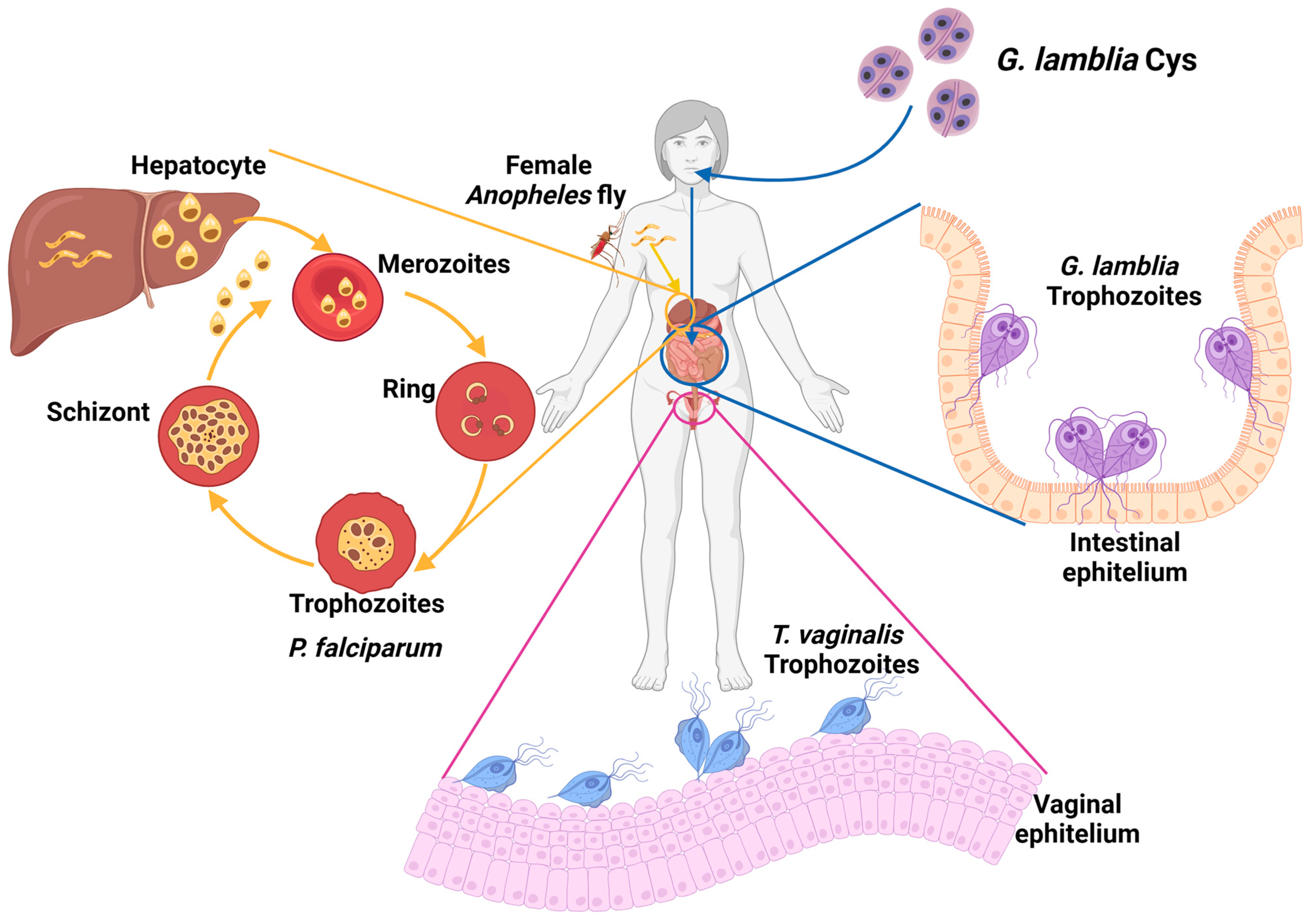
1. Parasitic Infections with an Impact on the Population
1.1. Giardiasis
1.2. Trichomoniases
1.3. Malaria
2. Treatment and Drug Resistance in Giardicidal, Trichomonicidal, and Antimalarial Therapies
2.1. Resistance to Nitroimidazole Drug Family
2.2. Resistance to Antimalarials
3. Search for New Targets in Drug Design
4. Carbohydrate Metabolism and Role of the Pentose Phosphate Pathway in Parasites
4.1. Glucose Metabolism
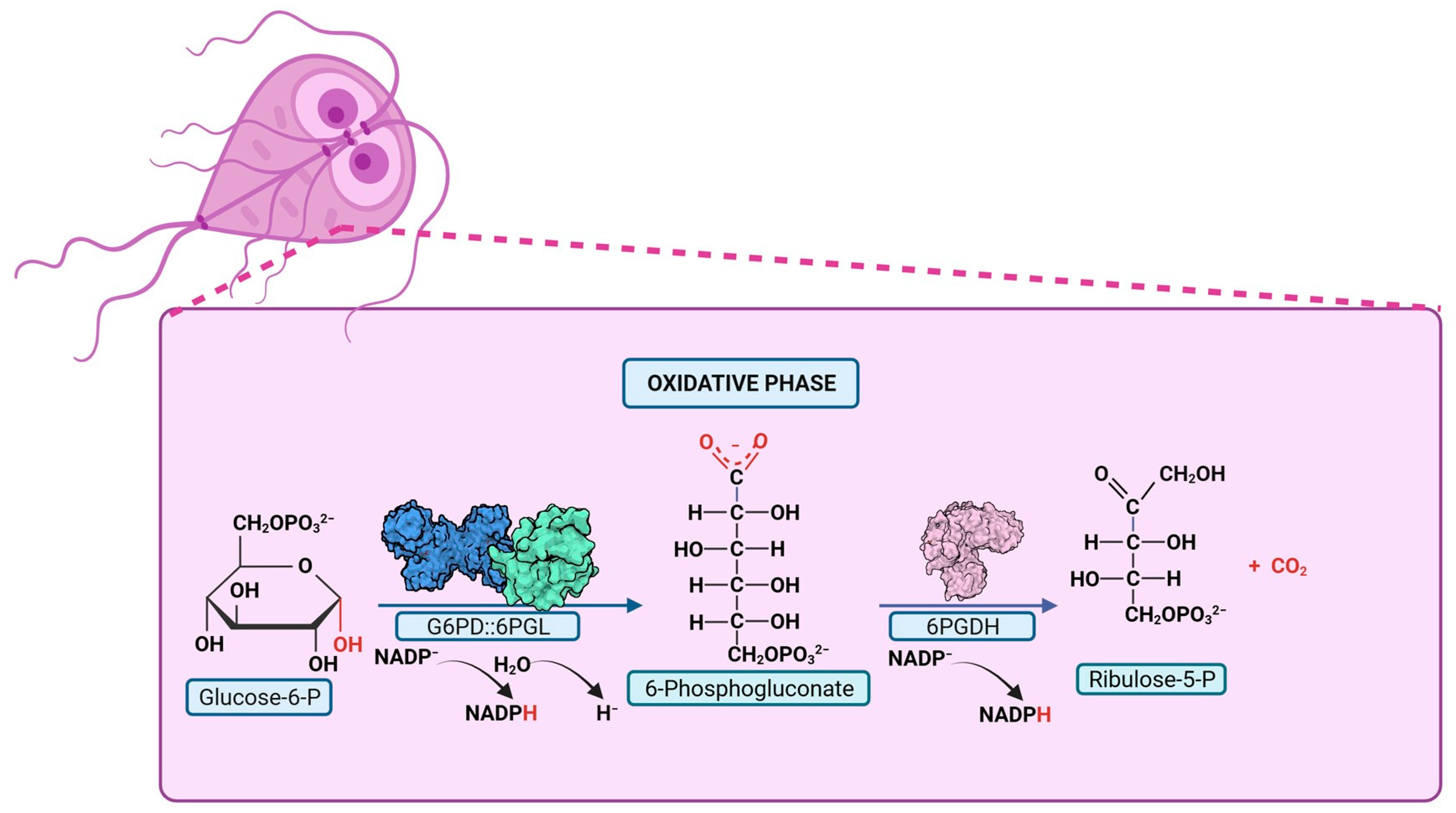
4.2. Pentose Phosphate Pathway: A Potential Origin of Therapeutic Targets
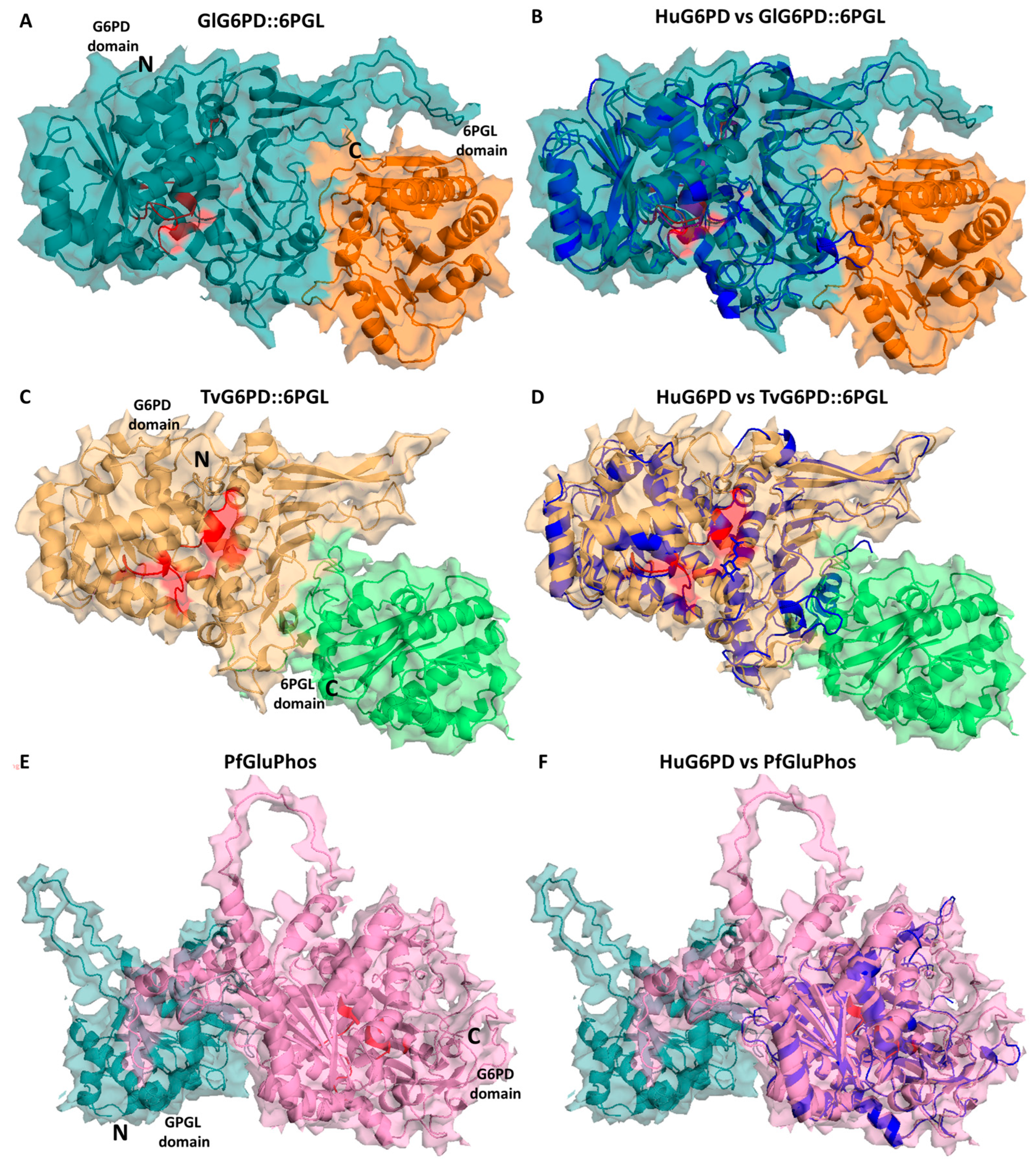
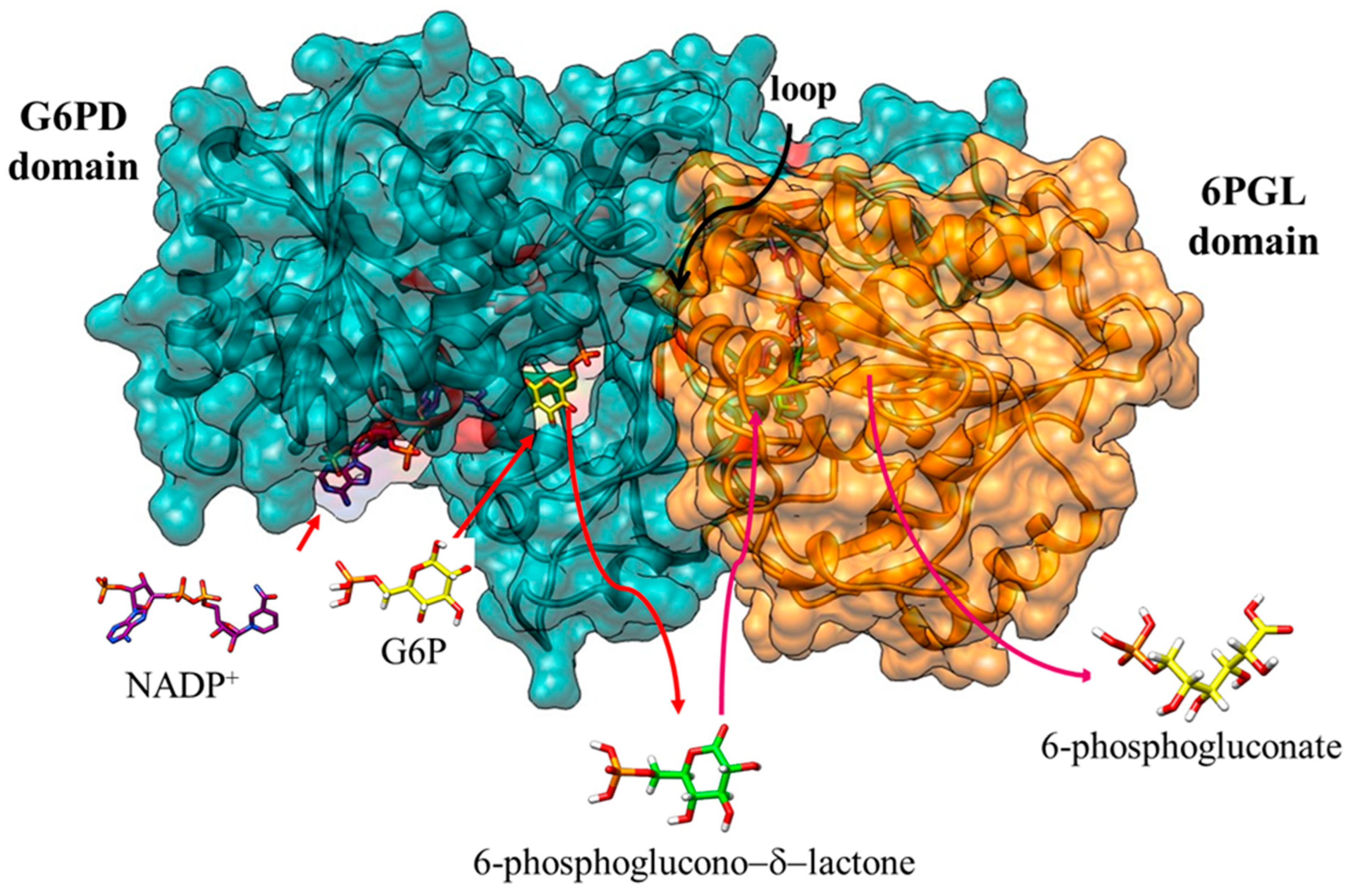
5. Structural and Kinetic Characteristic of the Bifunctional G6PD-6PGL Enzyme in Parasites
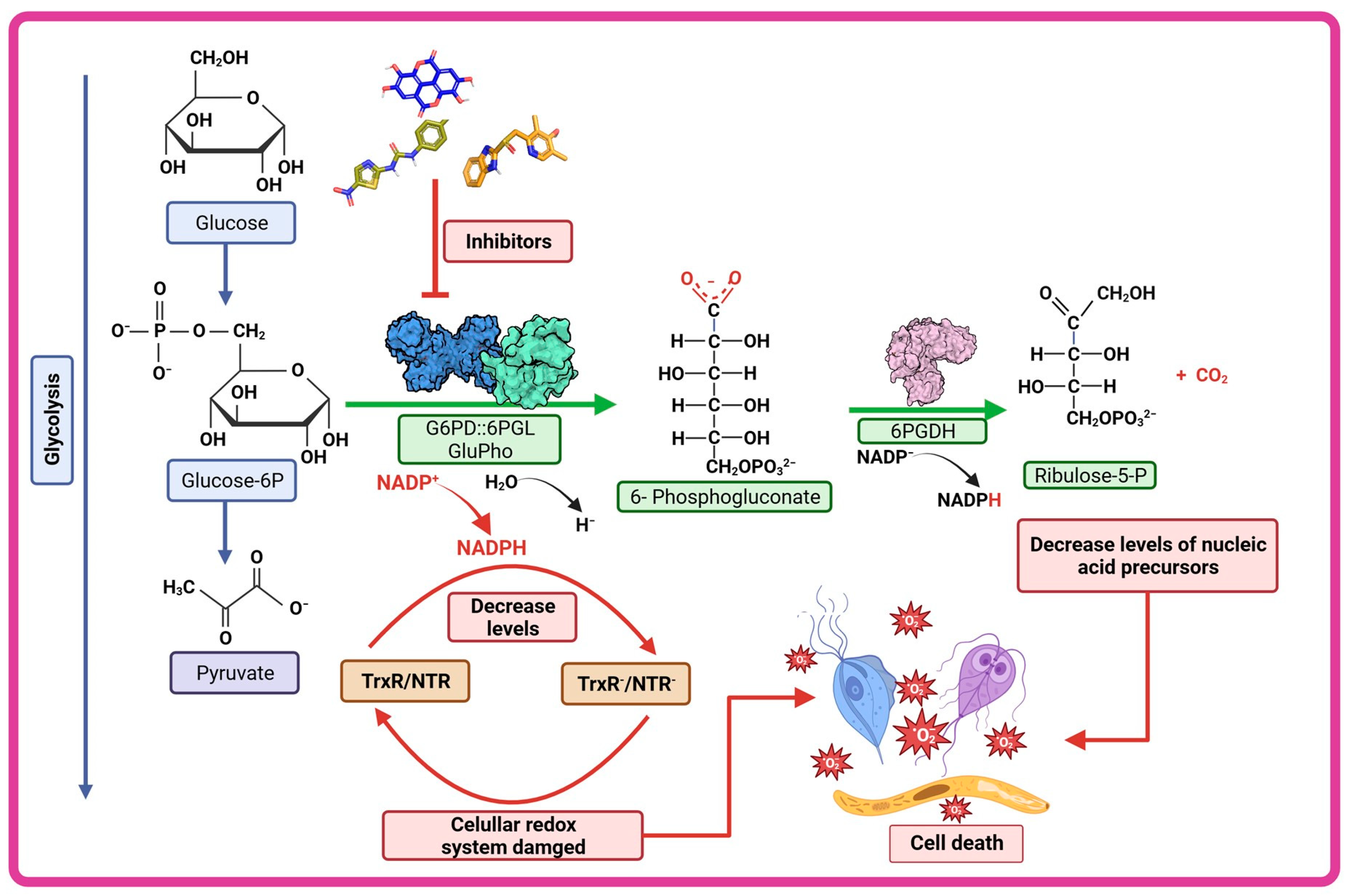
6. G6PD::6PGL Fused Enzyme in Parasites: Potential Drug Target
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Theel, E.S.; Pritt, B.S. Parasites. Microbiol. Spectr. 2016, 4, 411–466. [Google Scholar] [CrossRef]
- Ryan, U.M.; Feng, Y.; Fayer, R.; Xiao, L. Taxonomy and molecular epidemiology of Cryptosporidium and Giardia—A 50 year perspective (1971–2021). Int. J. Parasitol. 2021, 51, 1099–1119. [Google Scholar] [CrossRef]
- Edwards, T.; Burke, P.; Smalley, H.; Hobbs, G. Trichomonas vaginalis: Clinical relevance, pathogenicity and diagnosis. Crit. Rev. Microbiol. 2016, 42, 406–417. [Google Scholar] [CrossRef]
- Rowley, J.; Vander Hoorn, S.; Korenromp, E.; Low, N.; Unemo, M.; Abu-Raddad, L.J.; Chico, R.M.; Smolak, A.; Newman, L.; Gottlieb, S.; et al. Chlamydia, gonorrhoea, trichomoniasis and syphilis: Global prevalence and incidence estimates, 2016. Bull. World Health Organ. 2019, 97, 548–562. [Google Scholar] [CrossRef]
- Weinstock, H.; Berman, S.; Cates, W., Jr. Sexually transmitted diseases among American youth: Incidence and prevalence estimates, 2000. Perspect. Sex Reprod. Health 2004, 36, 6–10. [Google Scholar] [CrossRef]
- Shetty, A.K.; Steele, R.W. Plasmodium falciparum malaria. J. State Med. Soc. 1999, 151, 29–32. [Google Scholar]
- Walter, K.; John, C.C. Malaria. JAMA 2022, 327, 597. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.D. Giardia duodenalis: Biology and Pathogenesis. Clin. Microbiol. Rev. 2021, 34, e0002419. [Google Scholar] [CrossRef] [PubMed]
- Buret, A.G.; Cacciò, S.M.; Favennec, L.; Svärd, S. Update on Giardia: Highlights from the seventh International Giardia and Cryptosporidium Conference. Parasite 2020, 27, 49. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.Y.; Singer, S.M. The Intersection of Immune Responses, Microbiota, and Pathogenesis in Giardiasis. Trends Parasitol. 2017, 33, 901–913. [Google Scholar] [CrossRef]
- Carter, E.R.; Nabarro, L.E.; Hedley, L.; Chiodini, P.L. Nitroimidazole-refractory giardiasis: A growing problem requiring rational solutions. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2018, 24, 37–42. [Google Scholar] [CrossRef]
- Emery-Corbin, S.J.; Su, Q.; Tichkule, S.; Baker, L.; Lacey, E.; Jex, A.R. In vitro selection of Giardia duodenalis for Albendazole resistance identifies a β-tubulin mutation at amino acid E198K. Int. J. Parasitol. Drugs Drug Resist. 2021, 16, 162–173. [Google Scholar] [CrossRef]
- Loderstädt, U.; Frickmann, H. Antimicrobial resistance of the enteric protozoon Giardia duodenalis—A narrative review. Eur. J. Microbiol. Immunol. 2021, 11, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, M.; Bartelt, L.A. Giardia and growth impairment in children in high-prevalence settings: Consequence or co-incidence? Curr. Opin. Infect. Dis. 2022, 35, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Menezes, S.A.; Tasca, T. Essential Oils and Terpenic Compounds as Potential Hits for Drugs against Amitochondriate Protists. Trop. Med. Infect. Dis. 2023, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Report for Research on Infectious Diseases of Poverty; WHO: Geneva, Switzerland, 2012; pp. 1–184. ISBN 9789241564489.
- Hegazy, M.M.; El-Tantawy, N.L.; Soliman, M.M.; El-Sadeek, E.S.; El-Nagar, H.S. Performance of rapid immunochromatographic assay in the diagnosis of Trichomoniasis vaginalis. Diagn. Microbiol. Infect. Dis. 2012, 74, 49–53. [Google Scholar] [CrossRef]
- World Health Organization. World Health Statistics 2016: Monitoring Health for the SDGs, Sustainable Development Goals; World Health Organization: Geneva, Switzerland, 2018; ISBN 9789241565264.
- Crucitti, T.; Jespers, V.; Mulenga, C.; Khondowe, S.; Vandepitte, J.; Buvé, A. Non-sexual transmission of Trichomonas vaginalis in adolescent girls attending school in Ndola, Zambia. PLoS ONE 2011, 6, e16310. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.C.; Mthiyane, T.N.; Baisley, K.; Mchunu, S.L.; Ferguson, J.B.; Smit, T.; Crucitti, T.; Gareta, D.; Dlamini, S.; Mutevedzi, T.; et al. Prevalence of sexually transmitted infections among young people in South Africa: A nested survey in a health and demographic surveillance site. PLoS Med. 2018, 15, e1002512. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.P.; Feucht, U.D.; de Vos, L.; Ngwepe, P.; McIntyre, J.A.; Klausner, J.D.; Medina-Marino, A. Mother-to-child transmission of Chlamydia trachomatis, Neisseria gonorrhoeae, and Trichomonas vaginalis in HIV-infected pregnant women in South Africa. Int. J. STD AIDS 2021, 32, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Kissinger, P.J.; Gaydos, C.A.; Seña, A.C.; Scott McClelland, R.; Soper, D.; Secor, W.E.; Legendre, D.; Workowski, K.A.; Muzny, C.A. Diagnosis and Management of Trichomonas vaginalis: Summary of Evidence Reviewed for the 2021 Centers for Disease Control and Prevention Sexually Transmitted Infections Treatment Guidelines. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2022, 74, S152–S161. [Google Scholar] [CrossRef]
- Marques-Silva, M.; Lisboa, C.; Gomes, N.; Rodrigues, A.G. Trichomonas vaginalis and growing concern over drug resistance: A systematic review. J. Eur. Acad. Dermatol. Venereol. JEADV 2021, 35, 2007–2021. [Google Scholar] [CrossRef] [PubMed]
- Diéguez, I.S. Tricomoniasis: Una vision amplia. Latreia 2014, 27, 198–205. [Google Scholar]
- Workowski, K.A.; Bachmann, L.H.; Chan, P.A.; Johnston, C.M.; Muzny, C.A.; Park, I.; Reno, H.; Zenilman, J.M.; Bolan, G.A. Sexually Transmitted Infections Treatment Guidelines, 2021. MMWR Recomm. Rep. 2021, 70, 1–187. [Google Scholar] [CrossRef] [PubMed]
- Graves, K.J.; Novak, J.; Secor, W.E.; Kissinger, P.J.; Schwebke, J.R.; Muzny, C.A. A systematic review of the literature on mechanisms of 5-nitroimidazole resistance in Trichomonas vaginalis. Parasitology 2020, 147, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Daily, J.P. Update on pathogenesis, management, and control of Plasmodium vivax. Curr. Opin. Infect. Dis. 2022, 35, 404–409. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Report on Neglected Tropical Diseases 2023; WHO: Geneva, Switzerland, 2023; ISBN 9789240067295.
- World Health Organization. World Malaria Report 2022; WHO: Geneva, Switzerland, 2022; ISBN 9789240064898.
- Schiess, N.; Villabona-Rueda, A.; Cottier, K.E.; Huether, K.; Chipeta, J.; Stins, M.F. Pathophysiology and neurologic sequelae of cerebral malaria. Malar. J. 2020, 19, 266. [Google Scholar] [CrossRef] [PubMed]
- Escalante, A.A.; Cepeda, A.S.; Pacheco, M.A. Why Plasmodium vivax and Plasmodium falciparum are so different? A tale of two clades and their species diversities. Malar. J. 2022, 21, 139. [Google Scholar] [CrossRef] [PubMed]
- Su, X.-Z.; Zhang, C.; Joy, D.A. Host-Malaria Parasite Interactions and Impacts on Mutual Evolution. Front. Cell. Infect. Microbiol. 2020, 10, 587933. [Google Scholar] [CrossRef]
- Das, S.; Saha, B.; Hati, A.K.; Roy, S. Evidence of Artemisinin-Resistant Plasmodium falciparum Malaria in Eastern India. New Engl. J. Med. 2018, 379, 1962–1964. [Google Scholar] [CrossRef]
- Das, S.; Kar, A.; Manna, S.; Mandal, S.; Mandal, S.; Das, S.; Saha, B.; Hati, A.K. Artemisinin combination therapy fails even in the absence of Plasmodium falciparum kelch13 gene polymorphism in Central India. Sci. Rep. 2021, 11, 9946. [Google Scholar] [CrossRef]
- Fola, A.A.; Feleke, S.M.; Mohammed, H.; Brhane, B.G.; Hennelly, C.M.; Assefa, A.; Crudal, R.M.; Reichert, E.; Juliano, J.J.; Cunningham, J.; et al. Plasmodium falciparum resistant to artemisinin and diagnostics have emerged in Ethiopia. Nat. Microbiol. 2023, 8, 1911–1919. [Google Scholar] [CrossRef]
- Gamo, F.J.; Sanz, L.M.; Vidal, J.; de Cozar, C.; Alvarez, E.; Lavandera, J.L.; Vanderwall, D.E.; Green, D.V.; Kumar, V.; Hasan, S.; et al. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Guiguemde, W.A.; Shelat, A.A.; Bouck, D.; Duffy, S.; Crowther, G.J.; Davis, P.H.; Smithson, D.C.; Connelly, M.; Clark, J.; Zhu, F.; et al. Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Meister, S.; Plouffe, D.M.; Kuhen, K.L.; Bonamy, G.M.C.; Wu, T.; Barnes, S.W.; Bopp, S.E.; Borboa, R.; Bright, A.T.; Che, J.; et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 2011, 334, 1372–1377. [Google Scholar] [CrossRef]
- Ling, D.B.; Nguyen, W.; Looker, O.; Razook, Z.; McCann, K.; Barry, A.E.; Scheurer, C.; Wittlin, S.; Famodimu, M.T.; Delves, M.J.; et al. A Pyridyl-Furan Series Developed from the Open Global Health Library Block Red Blood Cell Invasion and Protein Trafficking in Plasmodium falciparum through Potential Inhibition of the Parasite’s PI4KIIIB Enzyme. ACS Infect. Dis. 2023, 9, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; Leung, A.A.; Wong, A.H.; Sergi, C.M.; Kam, J.K. Giardiasis: An Overview. Recent Patents Inflamm. Allergy Drug Discov. 2019, 13, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Escobedo, A.A.; Cimerman, S. Giardiasis: A pharmacotherapy review. Expert Opin. Pharmacother. 2007, 8, 1885–1902. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.M.; Dunn, L.A.; Upcroft, P.; Upcroft, J.A. Efficacy of antigiardial drugs. Expert Opin. Drug Saf. 2003, 2, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Graves, K.J.; Ghosh, A.P.; Schmidt, N.; Augostini, P.; Secor, W.E.; Schwebke, J.R.; Martin, D.H.; Kissinger, P.J.; Muzny, C.A. Trichomonas vaginalis Virus Among Women with Trichomoniasis and Associations with Demographics, Clinical Outcomes, and Metronidazole Resistance. Clin. Infect. Dis. 2019, 69, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Leitsch, D.; Kolarich, D.; Binder, M.; Stadlmann, J.; Altmann, F.; Duchêne, M. Trichomonas vaginalis: Metronidazole and other nitroimidazole drugs are reduced by the flavin enzyme thioredoxin reductase and disrupt the cellular redox system. Implications for nitroimidazole toxicity and resistance. Mol. Microbiol. 2009, 72, 518–536. [Google Scholar] [CrossRef]
- Leitsch, D.; Burgess, A.G.; Dunn, L.A.; Krauer, K.G.; Tan, K.; Duchene, M. Pyruvate:ferredoxin oxidoreductase and thioredoxin reductase are involved in 5-nitroimidazole activation while flavin metabolism is linked to 5-nitroimidazole resistance in Giar-dia lamblia. J. Antimicrob. Chemother. 2011, 66, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Leitsch, D.; Schlosser, S.; Burgess, A.; Duchêne, M. Nitroimidazole drugs vary in their mode of action in the human parasite Giardia lamblia. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Tejman-Yarden, N.; Millman, M.; Lauwaet, T.; Davids, B.J.; Gillin, F.D.; Dunn, L.; Upcroft, J.A.; Miyamoto, Y.; Eckmann, L. Impaired parasite attachment as fitness cost of metronidazole resistance in Giardia lamblia. Antimicrob. Agents Chemother. 2011, 55, 4643–4651. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Wastling, J.; Sanderson, S.; Müller, N.; Hemphill, A. A novel Giardia lamblia nitroreductase, GLNR1, interacts with nitazoxanide and other thiazolides. Antimicrob. Agents Chemother. 2007, 51, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Ley, S.; Felger, I.; Hemphill, A.; Müller, N. Identification of differentially expressed genes in a Giardia lamblia WB C6 clone resistant to nitazoxanide and metronidazole. J. Antimicrob. Chemother. 2008, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Schildknecht, P.; Müller, N. Metabolism of nitro drugs metronidazole and nitazoxanide in Giardia lamblia: Characterization of a novel nitroreductase (GlNR2). J. Antimicrob. Chemother. 2013, 68, 1781–1789. [Google Scholar] [CrossRef]
- Su, Q.; Baker, L.; Emery, S.; Balan, B.; Ansell, B.; Tichkule, S.; Mueller, I.; Svärd, S.G.; Jex, A. Transcriptomic analysis of albend-azole resistance in human diarrheal parasite Giardia duodenalis. Int. J. Parasitology. Drugs Drug Resist. 2023, 22, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Zhong, A.; Zhang, H.; Li, J. Insight into molecular diagnosis for antimalarial drug resistance of Plasmodium falciparum parasites: A review. Acta Trop. 2023, 241, 106870. [Google Scholar] [CrossRef]
- Valderramos, S.G.; Fidock, D.A. Transporters involved in resistance to antimalarial drugs. Trends Pharmacol. Sci. 2006, 27, 594–601. [Google Scholar] [CrossRef]
- von Seidlein, L.; Dondorp, A. Fighting fire with fire: Mass antimalarial drug administrations in an era of antimalarial resistance. Expert Rev. Anti. Infec. Ther. 2015, 13, 715–730. [Google Scholar] [CrossRef]
- Cowman, A.F.; Galatis, D.; Thompson, J.K. Selection for mefloquine resistance in Plasmodium falciparum is linked to am-plification of the pfmdr1 gene and crossresistance to halofantrine and quinine. Proc. Natl. Acad. Sci. USA 1994, 91, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Duraisingh, M.T.; Cowman, A.F. Contribution of the pfmdr1 gene to antimalarial drug-resistance. Acta Trop. 2005, 94, 181–190. [Google Scholar] [CrossRef]
- Takala-Harrison, S.; Clark, T.G.; Jacob, C.G.; Cummings, M.P.; Miotto, O.; Dondorp, A.M.; Fukuda, M.M.; Nosten, F.; Noedl, H.; Imwong, M.; et al. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc. Natl. Acad. Sci. USA 2013, 110, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Ariey, F.; Witkowski, B.; Amaratunga, C.; Beghain, J.; Langlois, A.C.; Khim, N.; Kim, S.; Duru, V.; Bouchier, C.; Ma, L. A mo-lecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Takala-Harrison, S.; Jacob, C.G.; Arze, C.; Cummings, M.P.; Silva, J.C.; Dondorp, A.M.; Fukuda, M.M.; Hien, T.T.; Mayxay, M.; Noedl, H.; et al. Independent Emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J. Infect. Dis. 2015, 211, 670–679. [Google Scholar] [CrossRef]
- Yang, T.; Yeoh, L.M.; Tutor, M.V.; Dixon, M.W.; McMillan, P.J.; Xie, S.C.; Bridgford, J.L.; Gillett, D.L.; Duffy, M.F.; Ralph, S.A.; et al. Decreased K13 Abundance Reduces Hemoglobin Catabolism and Proteotoxic Stress, Underpinning Artemisinin Resistance. Cell Rep. 2019, 29, 2917–2928.e5. [Google Scholar] [CrossRef]
- Atul; Chaudhary, P.; Gupta, S.; Shoaib, R.; Pasupureddy, R.; Goyal, B.; Kumar, B.; Singh, O.P.; Dixit, R.; Singh, S.; et al. Artemisinin resistance in P. falciparum: Probing the interacting partners of Kelch13 protein in parasite. J. Glob. Antimicrob. Resist. 2023, 35, 67–75. [Google Scholar] [CrossRef]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- Swinney, D.C. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin. Pharmacol. Ther. 2013, 93, 299–301. [Google Scholar] [CrossRef]
- Futamura, Y.; Muroi, M.; Osada, H. Target identification of small molecules based on chemical biology approaches. Mol. Biosyst. 2013, 9, 897–914. [Google Scholar] [CrossRef]
- Gilbert, I.H. Drug discovery for neglected diseases: Molecular target-based and phenotypic approaches. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ochoa, B.; Navarrete-Vázquez, G.; Nava-Zuazo, C.; Castillo-Villanueva, A.; Méndez, S.T.; Torres-Arroyo, A.; Gómez-Manzo, S.; Marcial-Quino, J.; Ponce-Macotela, M.; Rufino-González, Y.; et al. Novel giardicidal compounds bearing proton pump inhibitor scaffold proceeding through triosephosphate isomerase inactivation. Sci. Rep. 2017, 7, 7810. [Google Scholar] [CrossRef] [PubMed]
- Vique-Sánchez, J.L.; Caro-Gómez, L.A.; Brieba, L.G.; Benítez-Cardoza, C.G. Developing a new drug against trichomoniasis, new inhibitory compounds of the protein triosephosphate isomerase. Parasitol. Int. 2020, 76, 102086. [Google Scholar] [CrossRef]
- López-Velázquez, G.; Fernández-Lainez, C.; de la Mora-de la Mora, J.I.; Caudillo De La Portilla, D.; Reynoso-Robles, R.; González-Maciel, A.; Ridaura, C.; García-Torres, I.; Gutiérrez-Castrellón, P.; Olivos-García, A.; et al. On the molecular and cellular effects of omeprazole to further support its effectiveness as an antigiardial drug. Sci. Rep. 2019, 9, 8922. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Corrales, J.Á.; Josan, J.S. Proteomics for Drug Discovery: Methods and Protocols; Lazar, I.M., Kontoyianni, M., Lazar, A.C., Eds.; Springer: New York, NY, USA, 2017; pp. 207–219. [Google Scholar]
- Lindmark, D.G. Energy metabolism of the anaerobic protozoon Giardia lamblia. Mol. Biochem. Parasitol. 1980, 1, 1–12. [Google Scholar] [CrossRef]
- Upcroft, P.; Upcroft, J.A. Drug Targets and mechanisms of resistance in the anaerobic protozoa. Clin. Microbiol. Rev. 2001, 14, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Krieg, N.R.; Hoffman, P.S. Microaerophily and oxygen toxicity. Annu. Rev. Microbiol. 1986, 40, 107–130. [Google Scholar] [CrossRef]
- Lloyd, D.; Harris, J.C.; Biagini, G.A.; Hughes, M.R.; Maroulis, S.; Bernard, C.; Wadley, R.B.; Edwards, M.R. The plasma membrane of microaerophilic protists: Oxidative and nitrosative stress. Microbiology 2004, 150, 1183–1190. [Google Scholar] [CrossRef]
- Lloyd, D.; Coombs, G.H. Aerotolerantly anaerobic protozoa: Some unsolved problems. In Biochemistry and Molecular Biology of ‘Anaerobic’ Protozoa; Harwood Academic Publishers: Chur, Switzerland, 1989; pp. 267–286. [Google Scholar]
- Lloyd, D.; Harris, J.C.; Maroulis, S.; Biagini, G.A.; Wadley, R.B.; Turner, M.P.; Edwards, M.R. The microaerophilic flagellate Giardia intestinalis: Oxygen and its reaction products collapse membrane potential and cause cytotoxicity. Microbiology 2000, 146, 3109–3118. [Google Scholar] [CrossRef]
- Barrett, M. The pentose phosphate pathway and parasitic protozoa. Parasitol. Today 1997, 13, 11–16. [Google Scholar] [CrossRef]
- Dunn, A.M.; Hatcher, M.J. Parasites and biological invasions: Parallels, interactions, and control. Trends Parasitol. 2015, 31, 189–199. [Google Scholar] [CrossRef]
- Carvajal, C.C. Reactive oxygen species: Training, function and oxidative stress. Rev. Med. Leg. Costa Rica 2019, 36, 1. [Google Scholar]
- Sanz, M.; Lahoz, G.A.; Sosa, R.; Prieto, M. Introducción al sistema immune. Componentes celulares del sistema immune innato. Medicine 2017, 12, 1369–1378. [Google Scholar]
- Townson, S.M.; Upcroft, J.A.; Upcroft, P. Characterisation and purification of pyruvate: Ferredoxin oxidoreductase from Giardia duodenalis. Mol. Biochem. Parasitol. 1996, 79, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Schofield, P.; Costello, M.; Edwards, M.; O’Sullivan, W. The arginine dihydrolase pathway is present in Giardia intestinalis. Int. J. Parasitol. 1990, 20, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Schofield, P.J.; Edwards, M.R.; Matthews, J.; Wilson, J.R. The pathway of arginine catabolism in Giardia intestinalis. Mol. Biochem. Parasitol. 1992, 51, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Upcroft, J.A.; Edwards, M.R.; Upcroft, P. Anaerobic bacterial metabolism in the ancient eukaryote Giardia duodenalis. Int. J. Parasitol. 1998, 28, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Morales-Luna, L.; Hernández-Ochoa, B.; Ramírez-Nava, E.J.; Martínez-Rosas, V.; Ortiz-Ramírez, P.; Fernández-Rosario, F.; González-Valdez, A.; Cárdenas-Rodríguez, N.; Serrano-Posada, H.; Centeno-Leija, S.; et al. Characterizing the Fused TvG6PD::6PGL Protein from the Protozoan Trichomonas vaginalis, and Effects of the NADP+ Molecule on Enzyme Stability. Int. J. Mol. Sci. 2020, 21, 4831. [Google Scholar] [CrossRef]
- Wamelink, M.M.C.; Struys, E.A.; Jakobs, C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: A review. J. Inherit. Metab. Dis. 2008, 31, 703–717. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Morales-Luna, L.; González-Valdez, A.; Hernández-Ochoa, B.; Arreguin-Espinosa, R.; Ortega-Cuellar, D.; Castillo-Rodríguez, R.A.; Martínez-Rosas, V.; Cárdenas-Rodríguez, N.; Enríquez-Flores, S.; Canseco-Ávila, L.M.; et al. Glucose-6-Phosphate Dehydrogenase::6-Phosphogluconolactonase from the Parasite Giardia lamblia. A Molecular and Biochemical Perspective of a Fused Enzyme. Microorganisms 2021, 9, 1678. [Google Scholar] [CrossRef]
- Saltveit, M. Respiratory Metabolism. In The Commercial Storage of Fruits, Vegetables, and Florist and Nursery Stocks. Agriculture Handbook 66; Gross, K., Wang, C., Saltveit, M., Eds.; Agricultural Research Service, United States Department of Agriculture: Washington, DC, USA, 2016; pp. 68–74. [Google Scholar] [CrossRef]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, Z.; Zhu, Y.; Qin, S. Glucose-6-phosphate dehydrogenase: A biomarker and potential therapeutic target for cancer. Anti-Cancer Agents Med. Chem. 2014, 14, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Maleki, S.; Mærk, M.; Valla, S.; Ertesvåg, H. Mutational Analyses of Glucose Dehydrogenase and Glucose-6-Phosphate Dehydrogenase Genes in Pseudomonas fluorescens Reveal Their Effects on Growth and Alginate Production. Appl. Environ. Microbiol. 2015, 81, 3349–3356. [Google Scholar] [CrossRef] [PubMed]
- Storm, J.; Perner, J.; Aparicio, I.; Patzewitz, E.-M.; Olszewski, K.; Llinas, M.; Engel, P.C.; Müller, S. Plasmodium falciparum glutamate dehydrogenase a is dispensable and not a drug target during erythrocytic development. Malar. J. 2011, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Crooke, A.; Diez, A.; Mason, P.J.; Bautista, J.M. Transient silencing of Plasmodium falciparum bifunctional glu-cose-6-phosphate dehydrogenase-6-phosphogluconolactonase. FEBS J. 2006, 273, 1537–1546. [Google Scholar] [CrossRef]
- Christodoulou, D.; Kuehne, A.; Estermann, A.; Fuhrer, T.; Lang, P.; Sauer, U. Reserve Flux Capacity in the Pentose Phosphate Pathway by NADPH Binding Is Conserved across Kingdoms. iScience 2019, 19, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.L.; Sodeinde, O.; Mason, P.J. Glucose-6-phosphate dehydrogenase-6-phosphogluconolactonase. A novel bifunctional enzyme in malaria parasites. Eur. J. Biochem. 2001, 268, 2013–2019. [Google Scholar] [CrossRef]
- Morales-Luna, L.; Serrano-Posada, H.; González-Valdez, A.; Ortega-Cuellar, D.; Vanoye-Carlo, A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Rufino-González, Y.; Castillo-Rodríguez, R.A.; de la Cruz, V.P.; et al. Biochemical Characterization and Structural Modeling of Fused Glucose-6-Phosphate Dehydrogenase-Phosphogluconolactonase from Giardia lamblia. Int. J. Mol. Sci. 2018, 19, 2518. [Google Scholar] [CrossRef]
- O’Brien, E.; Kurdi-Haidar, B.; Wanachiwanawin, W.; Carvajal, J.L.; Vulliamy, T.J.; Cappadoro, M.; Mason, P.J.; Luzzatto, L. Cloning of the glucose 6-phosphate dehydrogenase gene from Plasmodium falciparum. Mol. Biochem. Parasitol. 1994, 64, 313–326. [Google Scholar] [CrossRef]
- Alencar, N.; Sola, I.; Linares, M.; Juárez-Jiménez, J.; Pont, C.; Viayna, A.; Vílchez, D.; Sampedro, C.; Abad, P.; Pérez-Benavente, S.; et al. First homology model of Plasmodium falciparum glucose-6-phosphate dehydrogenase: Discovery of selective substrate analog-based inhibitors as novel antimalarial agents. Eur. J. Med. Chem. 2018, 146, 108–122. [Google Scholar] [CrossRef]
- Hempelmann, E.; Wilson, R. Detection of glucose-6-phosphate dehydrogenase in malarial parasites. Mol. Biochem. Parasitol. 1981, 2, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Ling, I.T.; Wilson, R.J. Glucose-6-phosphate dehydrogenase activity of the malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 1988, 31, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Kurdi-Haidar, B.; Luzzatto, L. Expression and characterization of glucose-6-phosphate dehydrogenase of Plasmodium falciparum. Mol. Biochem. Parasitol. 1990, 41, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Shahabuddin, M.; Rawlings, D.J.; Kaslow, D.C. A novel glucose 6-phosphate-dehydrogenase in Plasmodium falciparum—cDNA and primary protein structure. Biochim. Biophys. Acta. 1994, 1219, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.L.; Sodeinde, O.; Mason, P.J. A unique insertion in Plasmodium berghei glucose-6-phosphate dehydrogenase-6-phosphogluconolactonase: Evolutionary and functional studies. Mol. Biochem. Parasitol. 2003, 127, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jortzik, E.; Mailu, B.M.; Preuss, J.; Fischer, M.; Bode, L.; Rahlfs, S.; Becker, K. Glucose-6-phosphate dehydrogen-ase-6-phosphogluconolactonase: A unique bifunctional enzyme from Plasmodium falciparum. Biochem. J. 2011, 436, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Terrón-Hernández, J.; De la Mora-De la Mora, I.; González-Valdez, A.; Marcial-Quino, J.; García-Torres, I.; Vanoye-Carlo, A.; López-Velázquez, G.; Hernández-Alcántara, G.; Oria-Hernández, J.; et al. The Stability of G6PD Is Affected by Mutations with Different Clinical Phenotypes. Int. J. Mol. Sci. 2014, 15, 21179–21201. [Google Scholar] [CrossRef]
- Stover, N.A.; Dixon, T.A.; Cavalcanti, A.R.O. Multiple independent fusions of glucose-6-phosphate dehydrogenase with enzymes in the pentose phosphate pathway. PLoS ONE 2011, 6, e22269. [Google Scholar] [CrossRef]
- Preuss, J.; Hedrick, M.; Sergienko, E.; Pinkerton, A.; Mangravita-Novo, A.; Smith, L.; Marx, C.; Fischer, E.; Jortzik, E.; Rahlfs, S.; et al. High-throughput screening for small-molecule inhibitors of Plasmodium falciparum glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase. J. Biomol. Screen. 2012, 17, 738–751. [Google Scholar] [CrossRef]
- Preuss, J.; Maloney, P.; Peddibhotla, S.; Hedrick, M.P.; Hershberger, P.; Gosalia, P.; Milewski, M.; Li, Y.L.; Sugarman, E.; Hood, B.; et al. Discovery of a Plasmodium falciparum glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase inhibitor (R,Z)-N-((1-ethylpyrrolidin-2-yl)methyl)-2-(2-fluorobenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxamide (ML276) that reduces parasite growth in vitro. J. Med. Chem. 2012, 55, 7262–7272. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, K.; Berneburg, I.; Jortzik, E.; Hahn, J.; Rahbari, M.; Schulz, N.; Preuss, J.; Zapol’skii, V.A.; Bode, L.; Pinkerton, A.B.; et al. Glucose 6-phosphate dehydrogenase 6-phosphogluconolactonase: Characterization of the Plasmodium vivax enzyme and inhibitor studies. Malar. J. 2019, 18, 22. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.M.; Lim, E.E.; Jortzik, E.; Preuss, J.; Chua, H.H.; MacRae, J.I.; Rahlfs, S.; Haeussler, K.; Downton, M.T.; McConville, M.J.; et al. Plasmodium falciparumglucose-6-phosphate dehydrogenase 6-phosphogluconolactonase is a potential drug target. FEBS J. 2015, 282, 3808–3823. [Google Scholar] [CrossRef] [PubMed]
- Maloney, P.; Hedrick, M.; Peddibhotla, S.; Hershberger, P.; Milewski, M.; Gosalia, P.; Li, L.; Preuss, J.; Sugarman, E.; Hood, B.; et al. A 2nd Selective Inhibitor of Plasmodium falciparum Glucose-6-Phosphate Dehydrogenase (PfG6PDH) Probe 2. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Berneburg, I.; Peddibhotla, S.; Heimsch, K.C.; Haeussler, K.; Maloney, P.; Gosalia, P.; Preuss, J.; Rahbari, M.; Skorokhod, O.; Valente, E.; et al. An Optimized Dihydrodibenzothiazepine Lead Compound (SBI-0797750) as a Potent and Selective Inhibitor of Plasmodium falciparum and P. vivax Glucose 6-Phosphate Dehydrogenase 6-Phosphogluconolactonase. Antimicrob. Agents Chemother. 2022, 66, e0210921. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ochoa, B.; Martínez-Rosas, V.; Morales-Luna, L.; Calderón-Jaimes, E.; Rocha-Ramírez, L.M.; Ortega-Cuellar, D.; Rufino-González, Y.; González-Valdez, A.; Arreguin-Espinosa, R.; Enríquez-Flores, S.; et al. Pyridyl Methylsulfinyl Benzimidazole Derivatives as Promising Agents against Giardia lamblia and Trichomonas vaginalis. Molecules 2022, 27, 8902. [Google Scholar] [CrossRef] [PubMed]
- Morales-Luna, L.; Hernández-Ochoa, B.; Martínez-Rosas, V.; Navarrete-Vázquez, G.; Ortega-Cuellar, D.; Rufino-González, Y.; González-Valdez, A.; Arreguin-Espinosa, R.; Franco-Vásquez, A.M.; de la Cruz, V.P.; et al. Giardia lamblia G6PD::6PGL Fused Protein Inhibitors Decrease Trophozoite Viability: A New Alternative against Giardiasis. Int. J. Mol. Sci. 2022, 23, 14358. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Rosas, V.; Hernández-Ochoa, B.; Navarrete-Vázquez, G.; Martínez-Conde, C.; Gómez-Chávez, F.; Morales-Luna, L.; González-Valdez, A.; Arreguin-Espinosa, R.; Enríquez-Flores, S.; de la Cruz, V.P.; et al. Kinetic and Molecular Docking Studies to Determine the Effect of Inhibitors on the Activity and Structure of Fused G6PD::6PGL Protein from Trichomonas vaginalis. Molecules 2022, 27, 1174. [Google Scholar] [CrossRef]
- Scior, T.; Lozano-Aponte, J.; Ajmani, S.; Hernández-Montero, E.; Chávez-Silva, F.; Hernández-Núñez, E.; Moo-Puc, R.; Fraguela-Collar, A.; Navarrete-Vázquez, G. Antiprotozoal Nitazoxanide Derivatives: Synthesis, Bioassays and QSAR Study Combined with Docking for Mechanistic Insight. Curr. Comput. Aided-Drug Des. 2015, 11, 21–31. [Google Scholar] [CrossRef]
- Colín-Lozano, B.; León-Rivera, I.; Chan-Bacab, M.J.; Ortega-Morales, B.O.; Moo-Puc, R.; López-Guerrero, V.; Hernández-Núñez, E.; Argüello-Garcia, R.; Scior, T.; Barbosa-Cabrera, E.; et al. Synthesis, in vitro and in vivo giardicidal activity of nitrothiazole-NSAID chimeras displaying broad antiprotozoal spectrum. Bioorganic Med. Chem. Lett. 2017, 27, 3490–3494. [Google Scholar] [CrossRef]
- Hernández-Ochoa, B.; Gómez-Manzo, S.; Sánchez-Carrillo, A.; Marcial-Quino, J.; Rocha-Ramírez, L.M.; Santos-Segura, A.; Ramírez-Nava, E.J.; Arreguin-Espinosa, R.; Cuevas-Cruz, M.; Méndez-Tenorio, A.; et al. Enhanced Antigiardial Effect of Omeprazole Analog Benzimidazole Compounds. Molecules 2020, 25, 3979. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Enzyme | Kinetic Parameters | Reference | |||
|---|---|---|---|---|---|---|
| Km G6P (µM) | Km NADP+ (µM) | Km 6PGL (µM) | kcat (s−1) | |||
| P. falciparum | G6PD | 33.2 | 6.1 | - | Nd | [104] |
| 6PGL | - | - | 172 | 106 | [104] | |
| PfGluPho | 19.2 | 6.5 | - | 8.6 | [104] | |
| G. lamblia | G6PD | 94.2 | 26.7 | - | 0.05 | [87] |
| 6PGL | - | - | 51.5 | 31.8 | [87] | |
| G6PD::6PGL | 18.1 | 13 | - | 31.8 | [96] | |
| T. vaginalis | G6PD | Nd | Nd | Nd | Nd | - |
| 6PGL | Nd | Nd | Nd | Nd | - | |
| G6PD::6PGL | 210 | 27 | - | 147 | [84] | |
| Homo sapiens | G6PD | 38.5 | 6.2 | - | 233 | [105] |
| 6PGL | - | - | 242 | 505215 | [104] | |
| Enzyme | Inhibitor | IC50 (µM) | k2 (M−1S−1) | Type Inhibition | Culture IC50 (µM) | Reference |
|---|---|---|---|---|---|---|
| PfGluPho | C276-1187 | 127 | ND | G6P: NC; NADP+ : NC | ND | [37,104] |
| CB22 | 4.6 | ND | G6P: NC; NADP+ : NC | ND | [107] | |
| CB61 | 5.1 | ND | G6P: MT; NADP+ : NC | ND | [107] | |
| CB62 | 11.6 | ND | ND | ND | [107] | |
| CB63 | 1.7 | ND | G6P: M; NADP+ : NC | ND | [107] | |
| CB64 | 19.9 | ND | ND | ND | [107] | |
| CB70 | 4.5 | ND | G6P: MT; NADP+ : NC | ND | [107] | |
| CB103 | 1.1 | ND | G6P: MT; NADP+ : NC | ND | [107] | |
| CB104 | 7.6 | ND | G6P: MT; NADP+ : NC | ND | [108] | |
| ML276 | 0.889 | ND | ND | 2.6 | [108] | |
| ML304 | 0.190 | ND | G6P: C; NADP+ : MT | 0.190 | [111] | |
| Ellagic acid | 0.076 | ND | G6P: MT; NADP+ : MT | 0.105 | [110] | |
| 21 (vz0527) | 1.72 | ND | ND | ND | [109] | |
| 4 (vz1732) | 0.9 | ND | G6P: NC; NADP+ : NC | ND | [109] | |
| SBI-0797750 | 0.0067 | ND | G6P: C; NADP+ : MT | 0.073 | [112] | |
| PvGluPho | Ellagic acid | 0.032 | ND | G6P: MT; NADP+ : MT | ND | [109] |
| ML304 | 15.3 | ND | G6P: C; NADP+ : MT | ND | [109] | |
| 21 (vz0527) | 0.2 | ND | ND | ND | [109] | |
| 4 (vz1732) | 0.2 | ND | ND | ND | [109] | |
| SBI-0797750 | 31 | ND | G6P: MT; NADP+ : MT | ND | [112] | |
| GlG6PD::6PGL | H-BZM-2 | 37 | 2.3 | ND | 36 | [113] |
| O2N-BZM-7 | 12 | 1.92 | ND | 14 | [113] | |
| O2N-BZM-9 | 20 | 6.13 | ND | 17 | [113] | |
| CNZ-7 | 150 | 0.97 | ND | 8.7 | [114] | |
| CNZ-8 | 80 | 0.60 | ND | 15 | [114] | |
| CMC-1 | 70 | 1.88 | ND | 15 | [114] | |
| FLP-2 | 256 | 0.82 | ND | 24.1 | [114] | |
| TvG6PD::6PGL | O2N-BZM-7 | 22 | 0.8 | ND | 6 | [113] |
| O2N-BZM-9 | 240 | 1.6 | ND | 4 | [113] | |
| CNZ-3 | 93 | 0.66 | G6P: NC; NADP+ : C | ND | [115] | |
| CNZ-17 | 356 | 0.38 | G6P: UC; NADP+ : UC | ND | [115] | |
| JMM-3 | 155 | 0.33 | G6P: UC; NADP+ : UC | ND | [115] | |
| MCC-7 | 260 | 0.26 | G6P: NC; NADP+ : UC | ND | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Luna, L.; Vázquez-Bautista, M.; Martínez-Rosas, V.; Rojas-Alarcón, M.A.; Ortega-Cuellar, D.; González-Valdez, A.; Pérez de la Cruz, V.; Arreguin-Espinosa, R.; Rodríguez-Bustamante, E.; Rodríguez-Flores, E.; et al. Fused Enzyme Glucose-6-Phosphate Dehydrogenase::6-Phosphogluconolactonase (G6PD::6PGL) as a Potential Drug Target in Giardia lamblia, Trichomonas vaginalis, and Plasmodium falciparum. Microorganisms 2024, 12, 112. https://doi.org/10.3390/microorganisms12010112
Morales-Luna L, Vázquez-Bautista M, Martínez-Rosas V, Rojas-Alarcón MA, Ortega-Cuellar D, González-Valdez A, Pérez de la Cruz V, Arreguin-Espinosa R, Rodríguez-Bustamante E, Rodríguez-Flores E, et al. Fused Enzyme Glucose-6-Phosphate Dehydrogenase::6-Phosphogluconolactonase (G6PD::6PGL) as a Potential Drug Target in Giardia lamblia, Trichomonas vaginalis, and Plasmodium falciparum. Microorganisms. 2024; 12(1):112. https://doi.org/10.3390/microorganisms12010112
Chicago/Turabian StyleMorales-Luna, Laura, Montserrat Vázquez-Bautista, Víctor Martínez-Rosas, Miriam Abigail Rojas-Alarcón, Daniel Ortega-Cuellar, Abigail González-Valdez, Verónica Pérez de la Cruz, Roberto Arreguin-Espinosa, Eduardo Rodríguez-Bustamante, Eden Rodríguez-Flores, and et al. 2024. "Fused Enzyme Glucose-6-Phosphate Dehydrogenase::6-Phosphogluconolactonase (G6PD::6PGL) as a Potential Drug Target in Giardia lamblia, Trichomonas vaginalis, and Plasmodium falciparum" Microorganisms 12, no. 1: 112. https://doi.org/10.3390/microorganisms12010112
APA StyleMorales-Luna, L., Vázquez-Bautista, M., Martínez-Rosas, V., Rojas-Alarcón, M. A., Ortega-Cuellar, D., González-Valdez, A., Pérez de la Cruz, V., Arreguin-Espinosa, R., Rodríguez-Bustamante, E., Rodríguez-Flores, E., Hernández-Ochoa, B., & Gómez-Manzo, S. (2024). Fused Enzyme Glucose-6-Phosphate Dehydrogenase::6-Phosphogluconolactonase (G6PD::6PGL) as a Potential Drug Target in Giardia lamblia, Trichomonas vaginalis, and Plasmodium falciparum. Microorganisms, 12(1), 112. https://doi.org/10.3390/microorganisms12010112