Abstract
Secondary metabolites are not essential for the growth of microorganisms, but they play a critical role in how microbes interact with their surroundings. In addition to this important ecological role, secondary metabolites also have a variety of agricultural, medicinal, and industrial uses, and thus the examination of secondary metabolism of plants and microbes is a growing scientific field. While the chemical production of certain secondary metabolites is possible, industrial-scale microbial production is a green and economically attractive alternative. This is even more true, given the advances in bioengineering that allow us to alter the workings of microbes in order to increase their production of compounds of interest. This type of engineering requires detailed knowledge of the “chassis” organism’s metabolism. Since the resources and the catalytic capacity of enzymes in microbes is finite, it is important to examine the tradeoffs between various bioprocesses in an engineered system and alter its working in a manner that minimally perturbs the robustness of the system while allowing for the maximum production of a product of interest. The in silico multi-objective analysis of metabolism using genome-scale models is an ideal method for such examinations.
1. Introduction
Secondary metabolites (a.k.a. idiolites) are small, structurally diverse, chemical compounds that are generated by plants and microbes. They are produced by secondary metabolic processes during the stationary phase (idiophase) of an organism’s lifecycle that follows its growth phase (trophophase). Although these compounds are not essential for microbial growth, they confer a selective advantage to the organisms in their environments and serve many diverse and important functions. These include roles in bacterial survival and ecological interactions. For example, it has been shown that certain diatoms excrete secondary metabolites to promote the growth of beneficial bacteria and encourage direct physical interaction with them, while dissuading attachment by opportunist organisms and hindering their growth [1].
Secondary metabolites are also economically and medicinally important. Many herbicides, fungicides, bio-insecticides, antibiotics, immunosuppressants, antitumor drugs, and other high-value bioactive compounds are byproducts of microbial secondary metabolism. For example, certain alkaloid secondary metabolites (e.g., Taxol) have anticancer functions and can be produced by a number of different fungi, but the yield significantly varies between the different organisms [2].
Secondary metabolites are usually difficult to synthesize chemically. But, at times, this method is preferred over harvesting from biological sources. This is because chemical synthesis avoids problems like variable product quality that are associated with some natural systems [3]. However, the complexity of certain secondary metabolites (e.g., most alkaloid molecules) often makes chemical synthesis difficult or impossible. For such compounds, bioproduction and extraction is the most economically practical strategy [4]; to improve the harvest yield, there are many efforts underway to maximize the rate and efficiency of the metabolic processes that produce these compounds. While in many cases the organisms that are used for the production of secondary metabolites already have the metabolic machinery needed to produce these compounds (e.g., many members of Actinomycetaceae), sometimes—due to a desire for an increased growth rate or a need to couple the production of the compounds to the synthesis of other biochemicals—microbial chassis organisms such as E. coli are engineered to produce these compounds. The latter process could be riskier than using naturally producing organisms because at times the secondary metabolite or a byproduct of its production can end up being toxic to the chassis organism.
2. Optimization of Microbial Production of Secondary Metabolites
Often, the natural rates of production of secondary metabolites are too low to be industrially profitable. Thus, metabolic engineers spend a lot of time optimizing the rate of production of these compounds. One way of ensuring that a compound of interest is produced optimally is to accelerate the activity of primary metabolic pathways that produce the precursors and reducing the equivalents that are needed for its production [5]. Simultaneously eliminating competing processes that could siphon away energy, nutrients, or enzymatic capacity from the production of secondary metabolites also improves the yield and rate of production.
Our ability to manipulate complex biosystems is continually improving and the field of synthetic biology has made significant advances during the last decade. We are using heterologous gene expression to add functions to chassis organisms for the production of secondary metabolites (e.g., [6,7,8,9,10]). We also design novel synthetic gene circuits that encode new biological behaviors, dynamics, and control in our engineered systems (e.g., [11,12,13]). However, our control is still limited, and even when using microbial platforms whose workings we know best, we are at the mercy of stochastic fluctuations and other nonlinear interactions with our synthetically engineered circuitry that make it difficult to be fully sure of engineering outcomes.
While it might be possible to pinpoint a handful of obvious competing processes intuitively in model organisms, this will not be possible for system-level engineering. This is because most of our intuitive ideas stem from reductionist ideas and studies. For example, reductionist biochemical thinking would suggest that the overexpression of “rate-limiting” enzymes should result in significant increases in the activity of certain desired processes and this would lead to the increased production of one or more compounds of interest. However, this is not the observed behavior. To identify all of the processes that affect secondary metabolite production, and to predict changes that would optimally improve production rate and yield, requires system-level analyses using computational tools. System-level analyses have shown that the control of metabolic fluxes is not a single biocomponent property but instead a network property that is distributed across many enzymes [14,15,16] and is dependent on the topology of a network.
Manipulating complex biosystems to do our bidding is not trivial. This is because oftentimes we are forcing a system to operate in a manner that contradicts the regulatory rules that evolution has set forth for it. These regulations ensure maximum fitness for a system in its ecological niche. We might not be able to overcome these regulations. But if we have some understanding of a system’s most important objectives and how it prioritizes them under different environmental conditions, then we can devise ways to either work with the system’s regulations or “bend” them as much as possible to optimize our engineering objectives. The metabolic systems of microorganisms, even well-studied model organisms like E. coli, are too complex to learn about such tradeoffs using only reductionist methods and targeted analyses. Comprehensive in silico analyses are needed to fully grasp the multifaceted workings of complex biosystems.
3. System-Level Computational Models of Metabolism
System-level computational analyses are possible today due to revolutionary advances in high-throughput analytical technologies that permit the rapid and facile collection of multiple types of system-level data [17,18,19]. There are many different types of models that use process-appropriate mathematical formalisms to examine the operating principles of bioprocesses and systems in general. Some of the most widely used and successful methods for examining metabolic processes at a system-level are constraint-based reconstruction and analysis (COBRA) methods. These methods utilize omics data, particularly annotated genomes, to reconstruct the metabolic pathways that exist in a biosystem. The annotated genome provides a list of enzymes that an organism can produce. Using this list of enzymes with information found in biochemical databases such as KEGG [20], ModelSEED [21], and MetaCyc [22], a list of reactions that can be catalyzed within a system are compiled. This list is used to reconstruct the metabolic pathways in an organism and serves as the basis for genome-scale metabolic models (GEMs) (see Figure 1). Currently there are several tools available that allow for the rapid generation of draft GEMs for organisms [23,24,25,26,27,28]. The build metabolic model app available on the U.S. Department of Energy’s systems biology knowledgebase (KBase) is one such tool [29]. One helpful advantage of using KBase for developing draft GEMs is the availability of a unique app (merge metabolic annotations) that allows for the import and combination of genome annotations from multiple sources. This app can greatly help the process of curating draft GEMs [30,31] by ensuring that the tool returns a more complete network reconstruction with few orphan gap-filled reactions [32].
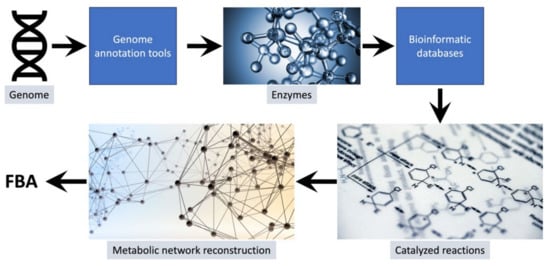
Figure 1.
The process for developing genome-scale models of metabolism. The genome of the organism is annotated using genome-annotation tools such as RAST [33], Prokka [34], or KOALA [35]. The annotated genome provides a list of enzymes that can be used with bioinformatic databases such as KEGG [20,36], ModelSEED [21], and MetaCyc [22] to generate a list of all the reactions that can occur in the organism at different times. This list when further curated with empirical data and information from the literature provides a reconstruction of the metabolic network of the organism that can be used for FBA and other types of COBRA modeling.
Detailed and accurate network reconstruction is particularly important for models that are used for studying organisms that produce secondary metabolites. This is because secondary and primary metabolism are tightly linked in these systems. The production of secondary metabolites requires precursor metabolites that are produced by primary metabolism. These include amino acids and short chain carboxylic acids. The precursors are used by proteins that are translated from groups of colocalized genes that work together to build the complex bioactive compounds. Polyketide synthases, non-ribosomal peptide synthases, and terpene cyclases are some of the most important classes of these biosynthetic gene clusters (BGCs). Most of the BGCs have characteristic catalytic domains that can be used to identify new gene clusters. This, along with the clustering characteristic of genes, has been used to develop computational tools (e.g., antiSMASH [37,38] and PRISM [39,40]) that can be used to identify BGCs from genome sequence data. These tools are critical for characterizing the secondary metabolism of organisms. The predicted biosynthetic capability can be used to build detailed GEMs that fully account for the connections between primary and secondary metabolism. The GEMs can then be used to identify the best candidate gene clusters for incorporation into engineered heterologous hosts.
3.1. Flux Balance Analysis
Flux balance analysis (FBA) is a widely used mathematical method for using GEMs to study metabolism in biosystems. FBA finds optimal metabolic steady-state activity fluxes that satisfy constraints imposed by the metabolic network structure, mass balance, and the availability of nutrients (see Figure 2). FBA uses the annotated genome-based metabolic network reconstruction of a system. The reconstruction is gap-filled using empirical data and fundamental biochemical and biophysical laws. The metabolic reactions are mathematically represented by the stoichiometric matrix, S (m × n), where m is the number of metabolites and n is the number of different reactions. FBA operates on assumptions of mass balance and metabolic steady state. Based on these assumptions, the following set of linear equations govern the system’s behavior:
where Xi is the concentration of metabolite i, Sij is the stoichiometry of metabolite i in reaction j, and νj is the flux of reaction j. Other constraints that are imposed on the model based on experimental measurements limit the amount of nutrients that a system can import and byproducts that are exported. Also, if information regarding the average concentration of an enzyme and its catalytic turnover rate are available, flux through a reaction can also be constrained. Thus, in properly bound FBA simulations all reactions have a lower and upper boundary:
where bi is the export/import flux of species i; α, β are the lower and upper bound for internal fluxes; and χ and φ are the lower and upper limits for transport fluxes. For FBA simulations it is assumed that cells are at steady state. Balanced growth is assumed and a biomass reaction with fixed stoichiometric values is added to the model. This reaction quantifies the amounts of various metabolites that are needed to produce 1 g of biomass.
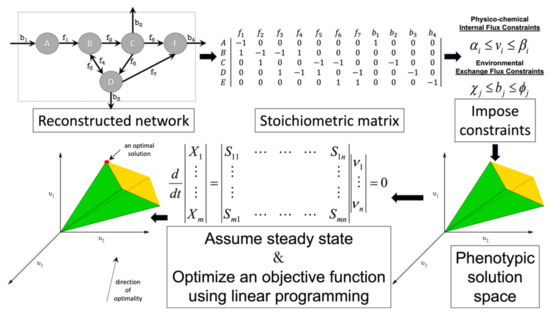
Figure 2.
Flux balance analysis (FBA) is the most widely used COBRA analysis method. The genome-based metabolic network reconstruction is converted to the mathematically useful format of a matrix that details the stoichiometry of all the reactions in the system. The model is constrained using empirical data and fundamental physico-chemical laws. The system is assumed to operate at steady state and linear programming is used to solve for a feasible flux pattern that optimizes the activity of one biological objective.
Once the GEM is appropriately constrained, FBA uses linear programming to solve for a feasible steady-state flux vector that optimizes an objective function. The most common objective function (cellular task to be optimized) is the production of biomass, i.e., cellular growth. However, other biological tasks can also be used as objective functions. There have been studies that have examined when and how such alternate objective functions might best be used [41,42]. Also, algorithms have been developed that use experimental data to infer the best objective functions for simulations [43,44]. Finally, as the use of FBA models expand into the realm of whole-cell modeling [45,46,47], variants of FBA have been introduced that loosen the rigid constraints imposed in classical FBA objective functions and bypass the limitations of steady-state and balanced growth assumptions [48].
FBA has been used to examine numerous biological topics that range from the fundamental nature of biological systems (e.g., [49,50,51]) to examining metabolism of deadly pathogens (e.g., [52,53,54,55]), studying cancer (e.g., [56,57,58,59]), elucidating the effects of genetic knockouts [60,61,62], and finding novel regulatory interactions [63]. The uses of FBA have continually increased. Automated tools for the generation of draft FBA models [21,23,24,25,26,27,28] have greatly helped this process. However, detailed analyses still require the use of human curated models. Even with advances in automated draft model generation, the overall process of developing high-fidelity models is still labor intensive [30,31].
Given the critical relationship between primary and secondary metabolism, system-level knowledge of the metabolic capabilities of an organism is critical for choosing the right biosystem for producing a secondary metabolite of interest. FBA studies are ideal tools for system-level analyses that can provide this information. In this vein, GEMs have been developed for several actinobacteria, particularly candidate members of the genus Streptomyces [64,65,66,67,68,69,70,71,72,73,74,75], a family of soil bacteria with diverse metabolisms that are known to produce a variety of different natural products, including anticancer drugs and antibiotics [76]. By using FBA with these GEMs, novel links between central metabolism and the production of secondary metabolites have been discovered. For example, it was found that the flux of nitrogen uptake and assimilation can positively affect the rate of antibiotic production in Streptomyces coelicolor [67,71]. Another study found that increasing pH can initiate the induction of idiophase in Streptomyces peucetius [66].
FBA has also been used to study the metabolic characteristics of the transition from active growth (trophophase) to idiophase. This transition is essential for the production of secondary metabolites because genes coding for the biosynthesis of secondary metabolites are usually not expressed at high growth rates [77]. The trophophase to idiophase transition is accompanied by an extensive rearrangement of cellular metabolism [72]. To gain a mechanistic understanding of these rearrangements, FBA was used to examine the flux patterns in the model secondary metabolite producer S. coelicolor during trophophase and idiophase [72]. FBA predicted that during the trophophase metabolites are mainly used for the production of biomass, while during idiophase the resources are shunted toward the production of secondary metabolites. The predicted flux pattern differences between the two phases were closely correlated with measured gene expression data. The correlation is so reliable that discrepancies between gene-expression data and predicted fluxes can be used to identify errors in genome annotation [72].
3.2. Multi-Objective Optimization
Optimizing the production and yield of a compound of interest usually requires engineering new strains of microbes. This is because natural selection has primed cellular metabolism for other biological objectives, such as growth and rapid adaptation to environmental pressures. The overproduction of compounds that are important to humans but have ancillary benefits for the biosystem are not prioritized. Cellular metabolism is tightly regulated to ensure against such “wasteful” overproduction of secondary metabolites. Therefore, efforts to overproduce secondary metabolites are often hindered by pathway competition with prioritized processes that are linked to the production of biomass components [78,79].
Given this divergence of biosystems’ and engineers’ objectives, engineering new idiolite-producing strains requires finding genetic manipulations that can work around the internal regulations of the systems while mitigating the fitness costs that may result from the induced perturbation. It is important to ensure that our engineered alterations do not drastically change the natural balance of biological objectives and subsequently the fitness of a system for a given environment.
GEMs can be powerful tools for identifying targets for such genetic manipulations. However, methods other than classical FBA need to be used. This is because FBA only optimizes one biological objective, while we need to examine the tradeoffs between multiple objectives. This is because evolution necessitates that organisms operate multiple critical processes at once and maximize the distribution of their limited resources in a Pareto-optimal fashion. A Pareto-optimal outcome is one where improvement in the performance of one task would result in diminishment of the ability to achieve (one or more) other tasks. To calculate Pareto-optimal solutions, examine the trade-offs between different objectives and identify bioprocesses that could hamper production or optimum yield of a desired product requires use of multi-objective (MO) optimization methods.
The multi-objective analysis of biological processes using constraint-based models is not a new method of analysis. One of the earliest such studies was the use of phenotype phase plane analysis (PPA) [80]. PPA was used to study the optimal uses of two model organisms’ metabolic networks as they adapt from variations of two environmental constraints [81,82]. Thus, PPA examined the tradeoffs between three system objectives (growth and the two constraints).
MO analyses are also preferred over plain FBA for optimizing the production of desired byproducts. Again, this is because FBA only optimizes one objective function, and unfortunately the usually optimized growth objective is often not appropriate for studying the production of natural products. For example, an analysis of the production of certain bioactive compounds by Streptomyces clavuligerus showed that the maximum ATP yield is the best objective function and that neither the maximization of growth nor bioactive compounds would agree with the experimental data [75]. If the objective that is optimized by FBA is growth, then the analysis will primarily examine the primary metabolism. It will ignore the secondary metabolism and production of compounds that are not biomass components. To overcome this shortcoming of FBA, several MO optimization methods have been developed to optimize the production of compounds of interest. We discuss some of them below.
3.2.1. OptKnock
One of the earliest MO optimization methods developed for strain engineering is OptKnock [83]. OptKnock is a bilevel optimization tool that uses GEMs and mixed-integer linear programing (MILP) to suggest gene manipulation strategies that result in the overproduction of a compound of interest. OptKnock achieves this by aligning the engineering objectives with the internal objectives of a biosystem. Thus, the proposed alterations work with the system instead of against it. OptKnock accounts for the fact that the metabolic flux distribution is controlled by system-specific internal objectives and that the surest way of ensuring a process is active is to make it essential for optimizing primary cellular objectives. To this end, OptKnock proposes genetic manipulations that make the production of the desired compound essential for cellular growth. For typical OptKnock simulations the maximization of biomass production (greater than a preset minimal level) is treated as the primal optimization while a dual optimization problem solves for reaction knockouts that maximize production of the compound of interest within the constraints set forth by optimization of primal problem (See Figure 3).
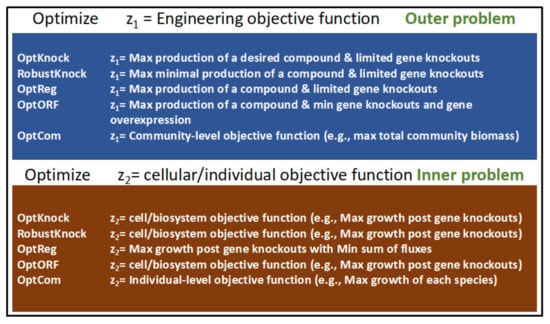
Figure 3.
A list of some bilevel optimization programs that have been developed for designing microbial strains that maximize the production of compounds of interest. The outer and inner optimization problems for each tool are listed.
OptKnock has been used for many strain design studies where the maximum production of a compound of interest was the goal (e.g., [84,85]). OptKnock has subsequently been updated with several variants. RobustKnock [86] expands upon OptKnock by identifying and eliminating competing pathways that could divert flux away from the production of compound of interest. OptReg [87] is a bilevel optimization platform that predicts the gene expression/enzyme level adjustments that could lead to the increased production of a compound of interest. OptORF [88] is another bilevel optimization platform that predicts engineering strategies using combinations of gene knockouts, differential gene expression, and the manipulation of regulatory pathways. OptFlux [89] is an open-source computational systems biology software that identifies engineering targets using evolutionary algorithms and simulated annealing. These meta-heuristic methods can work with different types of objective functions, including nonlinear ones. OptFlux also allows for the use of OptKnock for strain optimization.
3.2.2. OptStrain
OptStrain [90] is a major upgrade to OptKnock for designing new strains. This is because, unlike the other version of OptKnock, OptStrain can identify non-native genes/enzymes that can be incorporated into a system in order to increase the production of a desired compound. To inform OptStrain of the universal reactome, the developers of OptStrain compiled a regularly updated large database of biochemical reactions. OptStrain initially identifies a maximum yield pathway for the production of the desired compound from a selected substrate. This step is not species specific and uses all reactions deposited in the reaction database. OptStrain then uses combinatorial optimizations to search for stoichiometrically balanced pathways that include the minimum number of non-native reactions while satisfying the aim of maximizing the yield of the product. If GEMs are available for multiple host organisms, OptStrain can also be used to choose the best host for engineering projects. Once the set(s) of reactions/genes that need to be added to a system are identified, OptKnock can be used to predict the gene knockouts that will couple production of the compound of interest to cellular growth.
Bilevel optimization methods have been some of the most popular tools for strain engineering during the past two decades and are still being used for designing microbial strains that optimize the production of valuable products (e.g., [91,92]). Figure 3 lists some of the prominent bilevel optimization tools and their inner and outer objective functions.
3.2.3. MultiMetEval
MultiMetEval [93] is an MO optimization tool that utilizes COBRA methods to calculate the Pareto front between two cellular objectives. A Pareto front (see Figure 4) is a collection of Pareto-optimal solutions. The process of calculating a two-dimensional Pareto front (see Figure 4A,B) involves calculating the maximum () and minimum value of the first objective () using a GEM and FBA. The GEM is then updated with as a fixed value for the first objective. The updated model is then solved for the optimum value of the second objective (ϖ2,0) when the first objective operates at . This is one Pareto-optimal solution and one point on the Pareto front. If one wants to map the surface of the Pareto front using P points (including the one calculated at ), the GEM is constrained P − 1 times in an iterative fashion by the value:
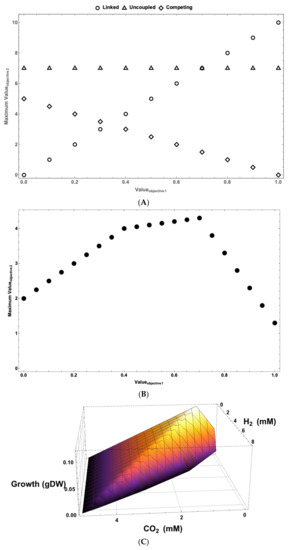
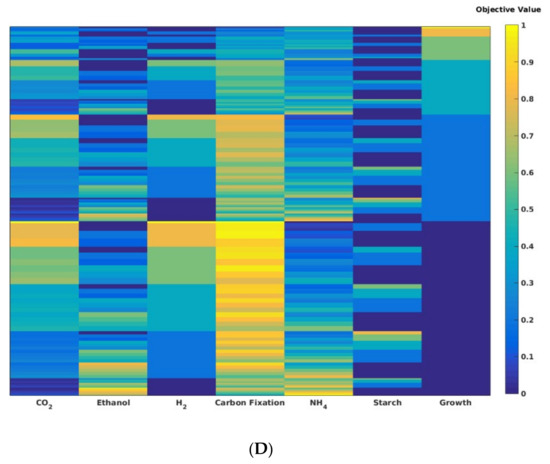
Figure 4.
Pareto fronts provide a wealth of information about the nature of interactions between different system objectives. (A) The 2D Pareto fronts show the nature of interactions between two system objectives. The interactions between objectives can be linked (improvement in one requires improvement in the other), uncoupled (value of one has no effect on the value of the other), or competing (increase in value of one lowers the value of the other). (B) In complex systems, the nature of interactions between two objectives can change depending on their values. Such multiphasic interactions can greatly help systems adapt to changes. Pareto fronts (depending on the number objectives that have been examined) can be visualized in a variety of different ways. (C) The 3D representation of a Pareto front can be used for visualizing the outcome of analysis from methods like PPA. This figure shows tradeoffs between hydrogen production, carbon fixation, and growth in Rhodopseudomonas palustris (based on results from Navid et al. [94]). (D) For analyses beyond three dimensions, heatmaps can be used to visualize the results. Here, a heatmap representing the Pareto front resulting from a seven-dimensional MOFA analysis of metabolism and biofuel production in Chlamydamonas reinhardtii is shown.
For each ϖ1,p value, the optimum value of the second objective (ϖ2,p) is calculated.
Two-dimensional Pareto fronts provide a wealth of information about the nature of interactions between two processes (objectives) in a system. As can be seen from Figure 4A, the interactions between objectives can be uncoupled, i.e., changes in the value of one would not affect the value of the other. They can be fully linked, i.e., any improvement in the operation of one requires an improvement in the value of the other. Alternatively, they could be competing, where any increase in the activity of one objective leads to a reduction in activity of the other. In most complex systems, the tradeoffs between objectives are not as simple as what is shown in Figure 4A. Instead, in most complex systems, the tradeoffs are multiphasic (see Figure 4B). This means that at different values for Objective 1, the nature of its interaction with Objective 2 changes. This could mean that the nature of interaction of an objective with another objective could change depending on its activity. For example, it could switch from being positively coupled to the other objective to competing with it (see the difference between Phases 2 and 3 in Figure 4B).
MultiMetEval also provides a platform to comparatively analyze metabolic differences between multiple GEMs. This can be used to identify organisms that are naturally adapted to overproducing secondary metabolites. The tool was used to examine the production capability of a large group of actinomycetes for different classes of secondary metabolite [93]. The study resulted in a number of interesting finds. One of the most intriguing finds was that organisms that have the most productive and metabolically versatile metabolisms are not the ones that are being used for biotechnology. Additionally, it was found that genome size and the complexity of secondary metabolism do not correlate with an organism’s productivity.
3.2.4. Multi-Objective Flux Analysis (MOFA)
While FBA studies typically optimize growth as the cell’s primary objective [42], it has been shown that no single objective fully governs the behavior of a system. A study examining the tradeoffs between double and triple combinations of various objectives in microbes showed that a Pareto-optimal combination of three primary tasks—maximum biomass yield, maximum yield of ATP, and the optimal allocation of resources—best describes the measured flux distribution for a variety of organisms and conditions [51]. However, the fluxes predicted for Pareto-optimal combinations of the primary objectives do not exactly match with experimental results. The Pareto optimization of other biological objectives that might be important for specific organisms and/or growth conditions could help to reduce the observed mismatch. The examination of these “secondary” objectives could also provide a quantitative measure of how these processes control the operation of a cell. This knowledge will help guide synthetic biology and metabolic engineering efforts so that the rates and yields of secondary metabolites are increased.
MO methods have been developed to study metabolic tradeoffs in systems that produce a variety of different high-value commodities such as biofuels (e.g., [95,96,97,98,99]). We recently developed a curated GEM for Rhodopseudomonas palustris [94], a metabolically versatile purple non-sulfur bacterium that has been studied as a model organism for the production of hydrogen gas (H2) [100,101,102]. To gain a better understanding of the tradeoffs between the different biological objectives (e.g., growth, ATP production, H2 production, carbon fixation, metabolism of aromatic compounds), we developed an MO analysis tool that we call multi-objective flux analysis (MOFA). Similar to Nagrath et al. [103], MOFA uses the normalized normal constraint (NNC) [104] method for MO analyses. This ensures that MOFA’s output is an n-dimensional (n = number of objectives) Pareto front comprised of a set of evenly distributed Pareto-optimal points, regardless of differences in the magnitudes of the examined objectives.
We developed MOFA because we wanted to examine the tradeoffs between more than a handful of objectives. Our high-dimensional (7 and 8 objectives) analyses provided us with novel insights into R. palustris’ ability to produce H2 as a biofuel. As with the production of secondary metabolites, we found that the production of H2 under all conditions drastically reduces the organism’s growth rate [94].
We have since developed a Matlab version of MOFA that can be used with the COBRA toolbox [105]. The COBRA toolbox is one of the most widely used platforms for computational systems-level analyses. We think this code would be useful for users of the COBRA toolbox who are interested in conducting high-dimensional MO analyses. Thus, we are including this code as a supplementary addition to this manuscript. A user guide is also included with the Supplementary Materials.
3.3. System-Level Analysis of Microbial Communities
Given that secondary metabolites have a major role in how microbes interact with their surroundings, engineering microbial communities can be one way of inducing the production of secondary metabolites [106,107,108,109]. Changing the dynamics of multicellular and multispecies systems by various means such as: altering the interactions between constituents via engineered loss or gain of functions, adding or reducing a member’s metabolic burden, or simply changing the community’s growth environment can result in changes in the secondary metabolism of individual organisms and the community as a whole. For example, one study has shown that dividing the metabolic pathways for the production of the chemotherapeutic compound paclitaxel improved its production [110]. As another example, it has been shown that in microbial cocultures presence of organisms that lower concentrations of metabolites that inhibit growth of another community member can greatly boost overall community growth rate [111]. In case of production of secondary metabolites, it has been shown that glucose interferes with biosynthesis of secondary metabolites [77]. So, addition of an organism that is a voracious consumer of glucose and producer of alternate forms of carbon could be a possible community engineering strategy for improving production of secondary metabolites.
3.3.1. OptCom
One MO-based tool developed for examining interactions in microbial communities is OptCom [112]. OptCom was developed by the same group that also developed OptKnock [83] and OptStrain [90]. As with these programs, OptCom utilizes bilevel optimization to examine the tradeoffs between individual vs. community fitness objectives in a multispecies microbial community. This allows for a quantitative and directed examination of the metabolic roles of each species in a community and the overall ecological niche. The nested bilevel optimization problem in OptCom is formulated using species-level objectives as the inner optimization while the community-level objective is optimized as the outer optimization (see Figure 3). Each species in the modeled community has its own biomass equation which is separately optimized in the inner level. The interactions between the constituent members of the community are constrained by limits on the exchange of metabolites between species. Community-level operation, such as the production of total community biomass, is optimized as the outer problem.
OptCom has been used to examine different types of interactions (positive, negative, or neutral) in microbial communities. This can be achieved by adjusting the inter-species flux constraints, i.e., varying the community-level optimization problem. A subsequent update to OptCom named d-OptCom [113] allowed for the simulation of transient changes in biomass of each community member. d-OptCom also allows changes to the concentrations of metabolites in the shared growth medium.
3.3.2. Community and Systems-Level Interactive Optimization (CASINO)
CASINO [114] is another bilevel optimization program that has been developed for the examination of interactions in complex multicellular and multispecies communities. CASINO uses community network properties to define the topology of a community. The inner problem is the optimization of biomass for individual species, while the outer problem is the optimization of biomass with the added aim of the optimum distribution of resources between species. The method was used to extensively study the interactions between the human gut microbiome and the host under different dietary regimens.
4. Conclusions
Genome-scale models of metabolism and COBRA methods have become indispensable tools for system-level analyses and metabolic engineering. COBRA methods have been used widely to study the metabolism of microbes. They have provided novel insights that have been used to engineer new microbial strains that have high production rates for commercially, industrially, and medicinally important bioactive compounds. For example, FBA simulation methods have been used with high-performance computing and pathway-search algorithms to predict putative heterologous biosynthesis pathways for over 6000 compounds in 70 different microbes [115].
While bilevel optimization tools and MO-based methods have been some of the most often-used tools for synthetic biology and metabolic engineering, other types of constraint-based modeling informed with different omics data could also contribute to these efforts. For example, it has been shown that metabolic flux changes associated with the transition of cellular metabolism from growth to idiophase closely correlate with gene expression dynamics. This provides a possible route for examining secondary metabolism in microbes by constraining GEMs with gene-expression data. Many tools have been developed for this purpose (e.g., [54,116,117,118,119,120,121,122]), and their uses and differences have been reviewed [123,124,125]. The deluge of heterogenous system-level data makes the use of these types of modeling essential for elucidating the metabolic state of a system under different conditions, particularly when the change in the environment is not biochemical but physical (e.g., temperature change). Beyond GEMs, the development and use of detailed whole-cell models [46,47] that account for the activity of every molecule in a system can greatly expand the role of computational models in the analysis of organisms capable of producing high-value bioactive compounds.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms11092149/s1. Code and manual for Matlab version of MOFA. References [104,105,126,127,128,129] are cited in the Supplementary Materials.
Author Contributions
Software, M.G.; supervision, A.N.; writing, A.N.; M.G. is deceased. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded as part of the LLNL’s μBiospheres Science Focus Area FWP SCW1039 supported by the Genome Sciences Program of the U.S. Department of Energy’s Office of Biological and Environmental Research.
Acknowledgments
This work is dedicated to the memory of Marc Griesemer, a wonderful friend and scientist. This work was carried out at Lawrence Livermore National Laboratory (LLNL) under Contract DE-AC52-07NA27344. LLNL-JRNL-852660.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the writing of the manuscript.
References
- Shibl, A.A.; Isaac, A.; Ochsenkuhn, M.A.; Cárdenas, A.; Fei, C.; Behringer, G.; Arnoux, M.; Drou, N.; Santos, M.P.; Gunsalus, K.C.; et al. Diatom modulation of select bacteria through use of two unique secondary metabolites. Proc. Natl. Acad. Sci. USA 2020, 117, 27445–27555. [Google Scholar] [CrossRef] [PubMed]
- Demain, A.L. Valuable Secondary Metabolites from Fungi. In Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites; Martín, J.-F., García-Estrada, C., Zeilinger, S., Eds.; Springer: New York, NY, USA, 2014; pp. 1–15. [Google Scholar]
- Smith, M.A.L. Large scale production of secondary metabolites. In Current Issues in Plant Molecular and Cellular Biology: Proceedings of the VIIIth International Congress on Plant Tissue and Cell Culture, Florence, Italy, 12–17 June, 1995; Terzi, M., Cella, R., Falavigna, A., Eds.; Springer: Berlin/Heidelberg, Germany, 1995; pp. 669–674. [Google Scholar]
- Murphy, D.J. Alkaloids. In Encyclopedia of Applied Plant Sciences, 2nd ed.; Thomas, B., Murray, B.G., Murphy, D.J., Eds.; Academic Press: Oxford, UK, 2017; pp. 118–124. [Google Scholar]
- Rokem, J.S.; Lantz, A.E.; Nielsen, J. Systems biology of antibiotic production by microorganisms. Nat. Prod. Rep. 2007, 24, 1262–1287. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Thodey, K.; Trenchard, I.; Smolke, C.D. Advancing secondary metabolite biosynthesis in yeast with synthetic biology tools. FEMS Yeast Res. 2012, 12, 144–170. [Google Scholar] [CrossRef] [PubMed]
- Beites, T.; Mendes, M.V. Chassis optimization as a cornerstone for the application of synthetic biology based strategies in microbial secondary metabolism. Front. Microbiol. 2015, 6, 906. [Google Scholar] [CrossRef]
- Rahmat, E.; Kang, Y. Yeast metabolic engineering for the production of pharmaceutically important secondary metabolites. Appl. Microbiol. Biotechnol. 2020, 104, 4659–4674. [Google Scholar] [CrossRef]
- Ruprecht, C.; Bönisch, F.; Ilmberger, N.; Heyer, T.V.; Haupt, E.T.K.; Streit, W.R.; Rabausch, U. High level production of flavonoid rhamnosides by metagenome-derived glycosyltransferase C in Escherichia coli utilizing dextrins of starch as a single carbon source. Metab. Eng. 2019, 55, 212–219. [Google Scholar] [CrossRef]
- Shrestha, A.; Pandey, R.P.; Dhakal, D.; Parajuli, P.; Sohng, J.K. Biosynthesis of flavone C-glucosides in engineered Escherichia coli. Appl. Microbiol. Biotechnol. 2018, 102, 1251–1267. [Google Scholar] [CrossRef]
- Baedeker, M.; Schulz, G.E. Overexpression of a designed 2.2 kb gene of eukaryotic phenylalanine ammonia-lyase in Escherichia coli. FEBS Lett. 1999, 457, 57–60. [Google Scholar] [CrossRef]
- Oßwald, C.; Zipf, G.; Schmidt, G.; Maier, J.; Bernauer, H.S.; Müller, R.; Wenzel, S.C. Modular construction of a functional artificial epothilone polyketide pathway. ACS Synth. Biol. 2014, 3, 759–772. [Google Scholar] [CrossRef]
- Yoshimura, T.; Shibata, N.; Hamano, Y.; Yamanaka, K. Heterologous production of hyaluronic acid in an ε-poly-L-lysine producer, Streptomyces albulus. Appl. Environ. Microbiol. 2015, 81, 3631–3640. [Google Scholar] [CrossRef]
- Teusink, B.; Bakker, B.M.; Westerhoff, H.V. Control of frequency and amplitudes is shared by all enzymes in three models for yeast glycolytic oscillations. Biochim. Biophys. Acta BBA Bioenerg. 1996, 1275, 204–212. [Google Scholar] [CrossRef]
- Kacser, H.; Burns, J.A. (Eds.) Rate control of biological processes. Symp. Soc. Exp. Biol. 1973, 27, 65. [Google Scholar]
- Groen, A.K.; Wanders, R.J.; Westerhoff, H.V.; van der Meer, R.; Tager, J.M. Quantification of the contribution of various steps to the control of mitochondrial respiration. J. Biol. Chem. 1982, 257, 2754–2757. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, T.D. Omics in Systems Biology: Current Progress and Future Outlook. Proteomics 2021, 21, 2000235. [Google Scholar] [CrossRef]
- Blencowe, M.; Arneson, D.; Ding, J.; Chen, Y.-W.; Saleem, Z.; Yang, X. Network modeling of single-cell omics data: Challenges, opportunities, and progresses. Emerg. Top. Life Sci. 2019, 3, 379–398. [Google Scholar]
- Cho, J.S.; Gu, C.; Han, T.H.; Ryu, J.Y.; Lee, S.Y. Reconstruction of context-specific genome-scale metabolic models using multiomics data to study metabolic rewiring. Curr. Opin. Syst. Biol. 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tisser, C.; et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008, 36, D623–D631. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef]
- Latendresse, M.; Ong, W.K.; Karp, P.D. Metabolic modeling with MetaFlux. In Microbial Systems Biology: Methods and Protocols; Humana Press: New York, NY, USA, 2022; pp. 259–289. [Google Scholar]
- Karlsen, E.; Schulz, C.; Almaas, E. Automated generation of genome-scale metabolic draft reconstructions based on KEGG. BMC Bioinform. 2018, 19, 467. [Google Scholar] [CrossRef]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef] [PubMed]
- Dias, O.; Rocha, M.; Ferreira, E.C.; Rocha, I. Reconstructing genome-scale metabolic models with merlin. Nucleic Acids Res. 2015, 43, 3899–3910. [Google Scholar] [CrossRef]
- Swainston, N.; Smallbone, K.; Mendes, P.; Kell, D.B.; Paton, N.W. The SuBliMinaL Toolbox: Automating steps in the reconstruction of metabolic networks. J. Integr. Bioinform. 2011, 8, 187–203. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States department of energy systems biology knowledgebase. Nat. Biotechnol. 2018, 36, 566. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Navid, A. Curating COBRA models of microbial metabolism. In Microbial Systems Biology: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2022; pp. 321–338. [Google Scholar]
- Griesemer, M.; Kimbrel, J.A.; Zhou, C.E.; Navid, A.; D’haeseleer, P. Combining multiple functional annotation tools increases coverage of metabolic annotation. BMC Genom. 2018, 19, 948. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32 (Suppl. S1), D277. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; Van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; De Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39 (Suppl. S2), W339–W346. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Dejong, C.A.; Rees, P.N.; Johnston, C.W.; Li, H.; Webster, A.L.H.; Wyatt, M.A.; Magarvey, N.A. Genomes to natural products prediction informatics for secondary metabolomes (PRISM). Nucleic Acids Res. 2015, 43, 9645–9662. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Merwin, N.J.; Johnston, C.W.; Magarvey, N.A. PRISM 3: Expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res. 2017, 45, W49–W54. [Google Scholar] [CrossRef]
- Schuetz, R.; Kuepfer, L.; Sauer, U. Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 2007, 3, 119. [Google Scholar] [CrossRef]
- Feist, A.M.; Palsson, B.O. The biomass objective function. Curr. Opin. Microbiol. 2010, 13, 344–349. [Google Scholar] [CrossRef]
- Burgard, A.P.; Maranas, C.D. Optimization-based framework for inferring and testing hypothesized metabolic objective functions. Biotechnol. Bioeng. 2003, 82, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Stettner, A.; Reznik, E.; Segrè, D.; Paschalidis, I.C. (Eds.) Learning cellular objectives from fluxes by inverse optimization. In Proceedings of the 2015 IEEE 54th Annual Conference on Decision and Control (CDC), Osaka, Japan, 15 December 2015; IEEE: Piscataway, NJ, USA. [Google Scholar]
- Karr, J.R.; Sanghvi, J.C.; Macklin, D.N.; Gutschow, M.V.; Jacobs, J.M.; Bolival, B.; Assad-Garcia, N.; Glass, J.I.; Covert, M.W. A whole-cell computational model predicts phenotype from genotype. Cell 2012, 150, 389–401. [Google Scholar] [CrossRef]
- Karr, J.R.; Takahashi, K.; Funahashi, A. The principles of whole-cell modeling. Curr. Opin. Microbiol. 2015, 27, 18–24. [Google Scholar] [CrossRef]
- Purcell, O.; Jain, B.; Karr, J.R.; Covert, M.W.; Lu, T.K. Towards a whole-cell modeling approach for synthetic biology. Chaos 2013, 23, 025112. [Google Scholar] [CrossRef] [PubMed]
- Birch, E.W.; Udell, M.; Covert, M.W. Incorporation of flexible objectives and time-linked simulation with flux balance analysis. J. Theor. Biol. 2014, 345, 12–21. [Google Scholar] [CrossRef]
- Almaas, E.; Kovacs, B.; Vicsek, T.; Oltvai, Z.N.; Barabasi, A.L. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature 2004, 427, 839–843. [Google Scholar] [CrossRef]
- Almaas, E.; Oltvai, Z.N.; Barabasi, A.L. The activity reaction core and plasticity of metabolic networks. PLoS Comput. Biol. 2005, 1, e68. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, R.; Zamboni, N.; Zampieri, M.; Heinemann, M.; Sauer, U. Multidimensional optimality of microbial metabolism. Science 2012, 336, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Navid, A.; Almaas, E. (Eds.) Genome-scale reconstruction of the metabolic network in Yersinia pestis CO922007. Mol. Biosyst. 2009, 5, 368–375. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chaudhury, S.; Abdulhameed, M.D.M.; Singh, N.; Tawa, G.J.; D’haeseleer, P.M.; Zemla, A.T.; Navid, A.; Zhou, C.E.; Franklin, M.C.; Cheung, J.; et al. Rapid countermeasure discovery against Francisella tularensis based on a metabolic network reconstruction. PLoS ONE 2013, 8, e63369. [Google Scholar] [CrossRef] [PubMed]
- Navid, A.; Almaas, E. Genome-level transcription data of Yersinia pestis analyzed with a New metabolic constraint-based approach. BMC Syst. Biol. 2012, 6, 150. [Google Scholar] [CrossRef]
- Presta, L.; Bosi, E.; Mansouri, L.; Dijkshoorn, L.; Fani, R.; Fondi, M. Constraint-based modeling identifies new putative targets to fight colistin-resistant A. baumannii infections. Sci. Rep. 2017, 7, 3706. [Google Scholar] [CrossRef]
- Folger, O.; Jerby, L.; Frezza, C.; Gottlieb, E.; Ruppin, E.; Shlomi, T. Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 2011, 7, 501. [Google Scholar] [CrossRef]
- Zielinski, D.C.; Jamshidi, N.; Corbett, A.J.; Bordbar, A.; Thomas, A.; Palsson, B.O. Systems biology analysis of drivers underlying hallmarks of cancer cell metabolism. Sci. Rep. 2017, 7, 41241. [Google Scholar] [CrossRef]
- Shlomi, T.; Benyamini, T.; Gottlieb, E.; Sharan, R.; Ruppin, E. Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Comput. Biol. 2011, 7, e1002018. [Google Scholar] [CrossRef] [PubMed]
- Asgari, Y.; Zabihinpour, Z.; Salehzadeh-Yazdi, A.; Schreiber, F.; Masoudi-Nejad, A. Alterations in cancer cell metabolism: The Warburg effect and metabolic adaptation. Genomics 2015, 105, 275–281. [Google Scholar] [CrossRef]
- Fong, S.S.; Palsson, B.O. Metabolic gene-deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat. Genet. 2004, 36, 1056–1058. [Google Scholar] [CrossRef]
- Segre, D.; Vitkup, D.; Church, G.M. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. USA 2002, 99, 15112–15117. [Google Scholar] [CrossRef]
- Shlomi, T.; Berkman, O.; Ruppin, E. Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc. Natl. Acad. Sci. USA 2005, 102, 7695–7700. [Google Scholar] [CrossRef]
- Herrgard, M.J.; Lee, B.S.; Portnoy, V.; Palsson, B.O. Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in Saccharomyces cerevisiae. Genome Res. 2006, 16, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Wen, J.; Wang, G.; Yu, G.; Jia, X.; Chen, Y. In silico aided metabolic engineering of Streptomyces roseosporus for daptomycin yield improvement. Appl. Microbiol. Biotechnol. 2012, 94, 637–649. [Google Scholar] [CrossRef]
- Medema, M.H.; Trefzer, A.; Kovalchuk, A.; van den Berg, M.; Müller, U.; Heijne, W.; Wu, L.; Alam, M.T.; Ronning, C.M.; Nierman, W.C.; et al. The Sequence of a 1.8-Mb bacterial linear plasmid reveals a rich evolutionary reservoir of secondary metabolic pathways. Genome Biol. Evol. 2010, 2, 212–224. [Google Scholar] [CrossRef]
- Kiviharju, K.; Moilanen, U.; Leisola, M.; Eerikäinen, T. A chemostat study of Streptomyces peucetius var. caesius N47. Appl. Microbiol. Biotechnol. 2007, 73, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Naeimpoor, F.; Mavituna, F. Metabolic Flux Analysis in Streptomyces coelicolor under Various Nutrient Limitations. Metab. Eng. 2000, 2, 140–148. [Google Scholar] [CrossRef]
- Borodina, I.; Siebring, J.; Zhang, J.; Smith, C.P.; van Keulen, G.; Dijkhuizen, L.; Nielsen, J. Antibiotic Overproduction in Streptomyces coelicolor A3(2) Mediated by Phosphofructokinase Deletion. J. Biol. Chem. 2008, 283, 25186–25199. [Google Scholar] [CrossRef] [PubMed]
- Borodina, I.; Krabben, P.; Nielsen, J. Genome-scale analysis of Streptomyces coelicolor A3 (2) metabolism. Genome Res. 2005, 15, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Laing, E.; Allenby, N.; Bucca, G.; Brenner, V.; Harrison, M.; Kierzek, A.M.; Smith, C.P. Metabolic and evolutionary insights into the closely-related species Streptomyces coelicolor and Streptomyces lividans deduced from high-resolution comparative genomic hybridization. BMC Genom. 2010, 11, 682. [Google Scholar] [CrossRef]
- Bum Kim, H.; Smith, C.P.; Micklefield, J.; Mavituna, F. Metabolic flux analysis for calcium dependent antibiotic (CDA) production in Streptomyces coelicolor. Metab. Eng. 2004, 6, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.T.; Merlo, M.E.; Hodgson, D.A.; Wellington, E.M.H.; Takano, E.; Breitling, R. Metabolic modeling and analysis of the metabolic switch in Streptomyces coelicolor. BMC Genom. 2010, 11, 202. [Google Scholar] [CrossRef]
- Gómez-Ríos, D.; López-Agudelo, V.A.; Ramírez-Malule, H.; Neubauer, P.; Junne, S.; Ochoa, S.; Ríos-Estepa, R. A Genome-scale insight into the effect of shear stress during the fed-batch production of clavulanic acid by Streptomyces clavuligerus. Microorganisms 2020, 8, 1255. [Google Scholar] [CrossRef]
- Kim, M.; Sang, Y.J.; Kim, J.; Kim, J.-N.; Kim, M.W.; Kim, B.-G. Reconstruction of a high-quality metabolic model enables the identification of gene overexpression targets for enhanced antibiotic production in Streptomyces coelicolor A3(2). Biotechnol. J. 2014, 9, 1185–1194. [Google Scholar] [CrossRef]
- Cavallieri, A.P.; Baptista, A.S.; Leite, C.A.; da Costa Araujo, M.L.G. A case study in flux balance analysis: Lysine, a cephamycin C precursor, can also increase clavulanic acid production. Biochem. Eng. J. 2016, 112, 42–53. [Google Scholar] [CrossRef]
- Harir, M.; Bendif, H.; Bellahcene, M.; Fortas, Z.; Pogni, R. Streptomyces secondary metabolites. Basic Biol. Appl. Actinobacteria 2018, 6, 99–122. [Google Scholar]
- Demain, A.L. Regulation of secondary metabolism in fungi. Pure Appl. Chem. 1986, 58, 219–226. [Google Scholar] [CrossRef]
- González-Lergier, J.; Broadbelt, L.J.; Hatzimanikatis, V. Theoretical considerations and computational analysis of the complexity in polyketide synthesis pathways. J. Am. Chem. Soc. 2005, 127, 9930–9938. [Google Scholar] [CrossRef] [PubMed]
- Paradise, E.M.; Kirby, J.; Chan, R.; Keasling, J.D. Redirection of flux through the FPP branch-point in Saccharomyces cerevisiae by down-regulating squalene synthase. Biotechnol. Bioeng. 2008, 100, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.S.; Ramakrishna, R.; Palsson, B.O. Characterizing the metabolic phenotype: A phenotype phase plane analysis. Biotechnol. Bioeng. 2002, 77, 27–36. [Google Scholar] [CrossRef]
- Edwards, J.S.; Ibarra, R.U.; Palsson, B.O. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotech. 2001, 19, 125–130. [Google Scholar] [CrossRef]
- Duarte, N.C.; Palsson, B.Ø.; Fu, P. Integrated analysis of metabolic phenotypes in Saccharomyces cerevisiae. BMC Genom. 2004, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. Optknock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 2003, 84, 647–657. [Google Scholar] [CrossRef]
- Pharkya, P.; Burgard, A.P.; Maranas, C.D. Exploring the overproduction of amino acids using the bilevel optimization framework OptKnock. Biotechnol. Bioeng. 2003, 84, 887–899. [Google Scholar] [CrossRef]
- Fong, S.S.; Burgard, A.P.; Herring, C.D.; Knight, E.M.; Blattner, F.R.; Maranas, C.D.; Palsson, B.O. In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol. Bioeng. 2005, 91, 643–648. [Google Scholar] [CrossRef]
- Tepper, N.; Shlomi, T. Predicting metabolic engineering knockout strategies for chemical production: Accounting for competing pathways. Bioinformatics 2010, 26, 536–543. [Google Scholar] [CrossRef]
- Pharkya, P.; Maranas, C.D. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab. Eng. 2006, 8, 1–13. [Google Scholar] [CrossRef]
- Kim, J.; Reed, J.L. OptORF: Optimal metabolic and regulatory perturbations for metabolic engineering of microbial strains. BMC Syst. Biol. 2010, 4, 53. [Google Scholar] [CrossRef]
- Rocha, I.; Maia, P.; Evangelista, P.; Vilaça, P.; Soares, S.; Pinto, J.P.; Nielsen, J.; Patil, K.R.; Ferreira, E.C.; Rocha, M. OptFlux: An open-source software platform for in silico metabolic engineering. BMC Syst. Biol. 2010, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Pharkya, P.; Burgard, A.P.; Maranas, C.D. OptStrain: A computational framework for redesign of microbial production systems. Genome Res. 2004, 14, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
- Purdy, H.M.; Pfleger, B.F.; Reed, J.L. Introduction of NADH-dependent nitrate assimilation in Synechococcus sp. PCC 7002 improves photosynthetic production of 2-methyl-1-butanol and isobutanol. Metab. Eng. 2022, 69, 87–97. [Google Scholar] [CrossRef]
- Malci, K.; Santibanez, R.; Jonguitud-Borrego, N.; Santoyo-Garcia, J.H.; Kherkoven, E.J.; Rios Solis, L. Improved Production of Taxol® Precursors in S. cerevisiae using Combinatorial in silico Design and Metabolic Engineering. bioRxiv 2023. bioRxiv:2023-06. [Google Scholar]
- Zakrzewski, P.; Medema, M.H.; Gevorgyan, A.; Kierzek, A.M.; Breitling, R.; Takano, E. MultiMetEval: Comparative and Multi-Objective Analysis of Genome-Scale Metabolic Models. PLoS ONE 2012, 7, e51511. [Google Scholar] [CrossRef]
- Navid, A.; Jiao, Y.; Wong, S.E.; Pett-Ridge, J. System-level analysis of metabolic trade-offs during anaerobic photoheterotrophic growth in Rhodopseudomonas palustris. BMC Bioinform. 2019, 20, 233. [Google Scholar] [CrossRef] [PubMed]
- Gowen, C.M.; Fong, S.S. Exploring Biodiversity for Cellulosic Biofuel Production. Chem. Biodivers. 2010, 7, 1086–1097. [Google Scholar] [CrossRef]
- Nazem-Bokaee, H.; Gopalakrishnan, S.; Ferry, J.G.; Wood, T.K.; Maranas, C.D. Assessing methanotrophy and carbon fixationfor biofuel production by Methanosarcina acetivorans. Microb. Cell Factories 2016, 15, 10. [Google Scholar] [CrossRef]
- Dash, S.; Mueller, T.J.; Venkataramanan, K.P.; Papoutsakis, E.T.; Maranas, C.D. Capturing the response of Clostridium acetobutylicum to chemical stressors using a regulated genome-scale metabolic model. Biotechnol. Biofuels 2014, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Shabestary, K.; Hudson, E.P. Computational metabolic engineering strategies for growth-coupled biofuel production by Synechocystis. Metab. Eng. Commun. 2016, 3, 216–226. [Google Scholar] [CrossRef]
- Fatma, Z.; Hartman, H.; Poolman, M.G.; Fell, D.A.; Srivastava, S.; Shakeel, T.; Yazdani, S.S. Model-assisted metabolic engineering of Escherichia coli for long chain alkane and alcohol production. Metab. Eng. 2018, 46, 1–12. [Google Scholar] [CrossRef]
- McKinlay, J.B.; Harwood, C.S. Photobiological production of hydrogen gas as a biofuel. Curr. Opin. Biotechnol. 2010, 21, 244–251. [Google Scholar] [CrossRef]
- Huang, J.J.; Heiniger, E.K.; McKinlay, J.B.; Harwood, C.S. Production of hydrogen gas from light and the inorganic electron donor thiosulfate by Rhodopseudomonas palustris. Appl. Environ. Microbiol. 2010, 76, 7717–7722. [Google Scholar] [CrossRef]
- Oh, Y.-K.; Seol, E.-H.; Kim, M.-S.; Park, S. Photoproduction of hydrogen from acetate by a chemoheterotrophic bacterium Rhodopseudomonas palustris P4. Int. J. Hydrogen Energy 2004, 29, 1115–1121. [Google Scholar] [CrossRef]
- Nagrath, D.; Avila-Elchiver, M.; Berthiaume, F.; Tilles, A.W.; Messac, A.; Yarmush, M.L. Soft Constraints-Based Multiobjective Framework for Flux Balance Analysis. Metab. Eng. 2010, 12, 429. [Google Scholar] [CrossRef] [PubMed]
- Messac, A.; Ismail-Yahaya, A.; Mattson, C.A. The normalized normal constraint method for generating the Pareto frontier. Struct. Multidiscip. Optim. 2003, 25, 86–98. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v. 3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef]
- Shurin, J.B.; Abbott, R.L.; Deal, M.S.; Kwan, G.T.; Litchman, E.; McBride, R.C.; Mandal, S.; Smith, V.H. Industrial-strength ecology: Trade-offs and opportunities in algal biofuel production. Ecol. Lett. 2013, 16, 1393–1404. [Google Scholar] [CrossRef]
- Lindemann, S.R.; Bernstein, H.C.; Song, H.-S.; Fredrickson, J.K.; Fields, M.W.; Shou, W.; Johnson, D.R.; Beliaev, A.S. Engineering microbial consortia for controllable outputs. ISME J. 2016, 10, 2077–2084. [Google Scholar] [CrossRef]
- Podolsky, I.A.; Seppälä, S.; Lankiewicz, T.S.; Brown, J.L.; Swift, C.L.; O’Malley, M.A. Harnessing nature’s anaerobes for biotechnology and bioprocessing. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 105–128. [Google Scholar] [CrossRef]
- Swift, C.L.; Brown, J.L.; Seppälä, S.; O’Malley, M.A. Co-cultivation of the anaerobic fungus Anaeromyces robustus with Methanobacterium bryantii enhances transcription of carbohydrate active enzymes. J. Ind. Microbiol. Biotechnol. 2019, 46, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Qiao, K.; Edgar, S.; Stephanopoulos, G. Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat. Biotechnol. 2015, 33, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Navid, A.; Stewart, B.; McKinlay, J.; Thelen, M.; Pett-Ridge, J. Syntrophic metabolism of a co-culture containing Clostridium cellulolyticum and Rhodopseudomonas palustris for hydrogen production. Int. J. Hydrogen Energy 2012, 37, 11719–11726. [Google Scholar] [CrossRef]
- Zomorrodi, A.R.; Maranas, C.D. OptCom: A multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput. Biol. 2012, 8, e1002363. [Google Scholar] [CrossRef] [PubMed]
- Zomorrodi, A.R.; Islam, M.M.; Maranas, C.D. d-OptCom: Dynamic multi-level and multi-objective metabolic modeling of microbial communities. ACS Synth. Biol. 2014, 3, 247–257. [Google Scholar] [CrossRef]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; De Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J.; et al. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef]
- Ding, S.; Cai, P.; Yuan, L.; Tian, Y.; Tu, W.; Zhang, D.; Cheng, X.; Sun, D.; Chen, J.; Hu, Q.N. CF-Targeter: A rational biological cell factory targeting platform for biosynthetic target chemicals. ACS Synth. Biol. 2019, 8, 2280–2286. [Google Scholar] [CrossRef]
- Zur, H.; Ruppin, E.; Shlomi, T. iMAT: An integrative metabolic analysis tool. Bioinformatics 2010, 26, 3140–3142. [Google Scholar] [CrossRef]
- Becker, S.A.; Palsson, B.O. Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.A.; Papin, J.A. Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 2011, 27, 541–547. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Price, N.D. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wallqvist, A.; Reifman, J. Modeling phenotypic metabolic adaptations of Mycobacterium tuberculosis H37Rv under hypoxia. PLoS Comput. Biol. 2012, 8, e1002688. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Pescini, D.; Galuzzi, B.G.; Bonanomi, M.; Gaglio, D.; Mangano, E.; Consolandi, C.; Alberghina, L.; Vanoni, M.; Damiani, C. INTEGRATE: Model-based multi-omics data integration to characterize multi-level metabolic regulation. PLoS Comput. Biol. 2022, 18, e1009337. [Google Scholar] [CrossRef]
- Hadadi, N.; Pandey, V.; Chiappino-Pepe, A.; Morales, M.; Gallart-Ayala, H.; Mehl, F.; Ivanisevic, J.; Sentchilo, V.; Meer, J.R.V.D. Mechanistic insights into bacterial metabolic reprogramming from omics-integrated genome-scale models. NPJ Syst. Biol. Appl. 2020, 6, 1. [Google Scholar] [CrossRef]
- Machado, D.; Herrgård, M. Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of eetabolism. PLoS Comput. Biol. 2014, 10, e1003580. [Google Scholar] [CrossRef]
- Cruz, F.; Faria, J.P.; Rocha, M.; Rocha, I.; Dias, O. A review of methods for the reconstruction and analysis of integrated genome-scale models of metabolism and regulation. Biochem. Soc. Trans. 2020, 48, 1889–1903. [Google Scholar] [CrossRef]
- Wendering, P.; Nikoloski, Z. Model-driven insights into the effects of temperature on metabolism. Biotechnol. Adv. 2023, 67, 108203. [Google Scholar] [CrossRef]
- Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2. 0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef]
- Becker, S.A.; Feist, A.M.; Mo, M.L.; Hannum, G.; Palsson, B.O.; Herrgard, M.J. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox. Nat. Protoc. 2007, 2, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Schilling, C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B.Ø. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 121. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).