
Metallo-Beta-Lactamase-like Encoding Genes in Candidate Phyla Radiation: Widespread and Highly Divergent Proteins with Potential Multifunctionality


 ,
,  ,
, 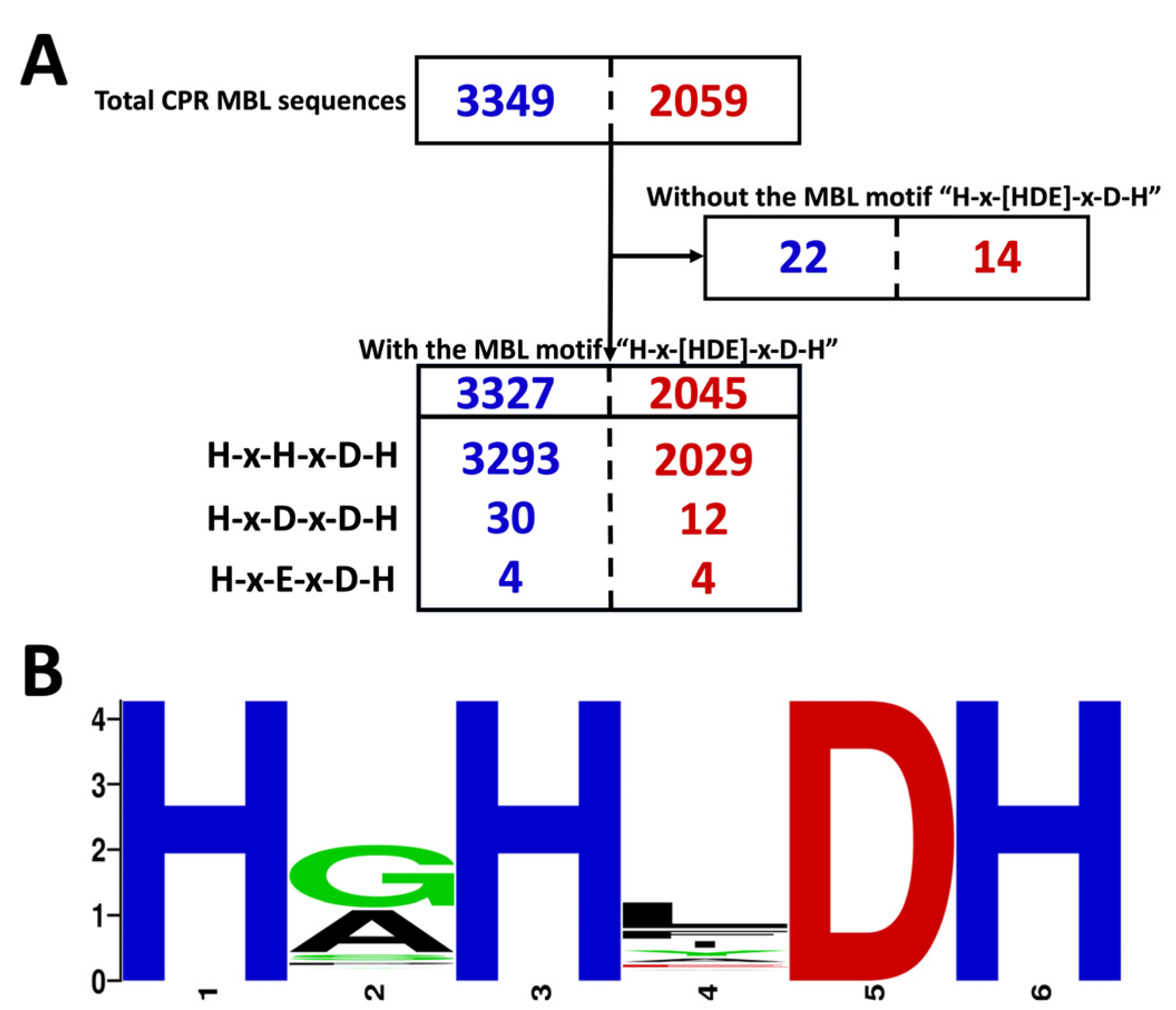{kind=link}
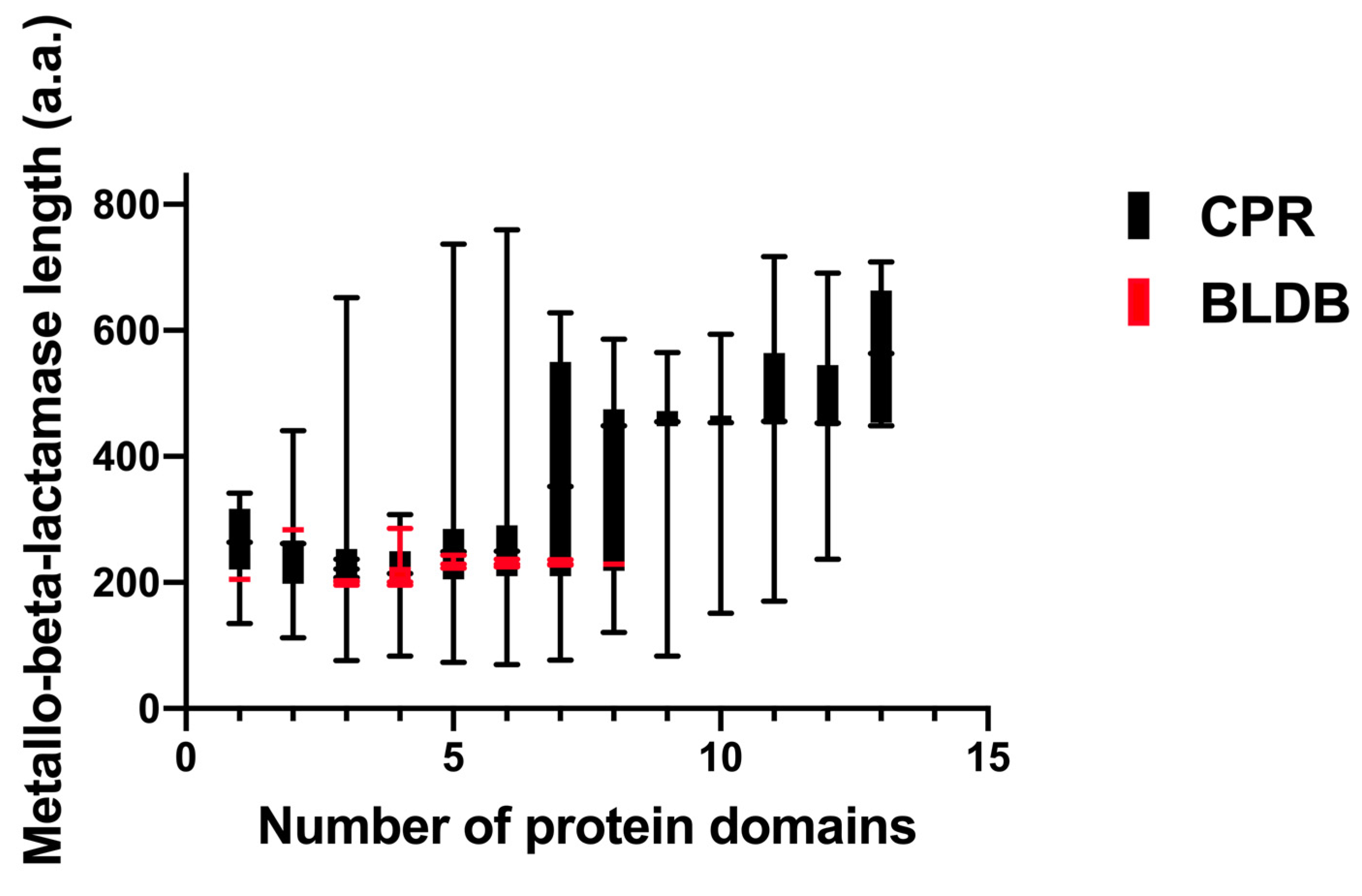{kind=link}
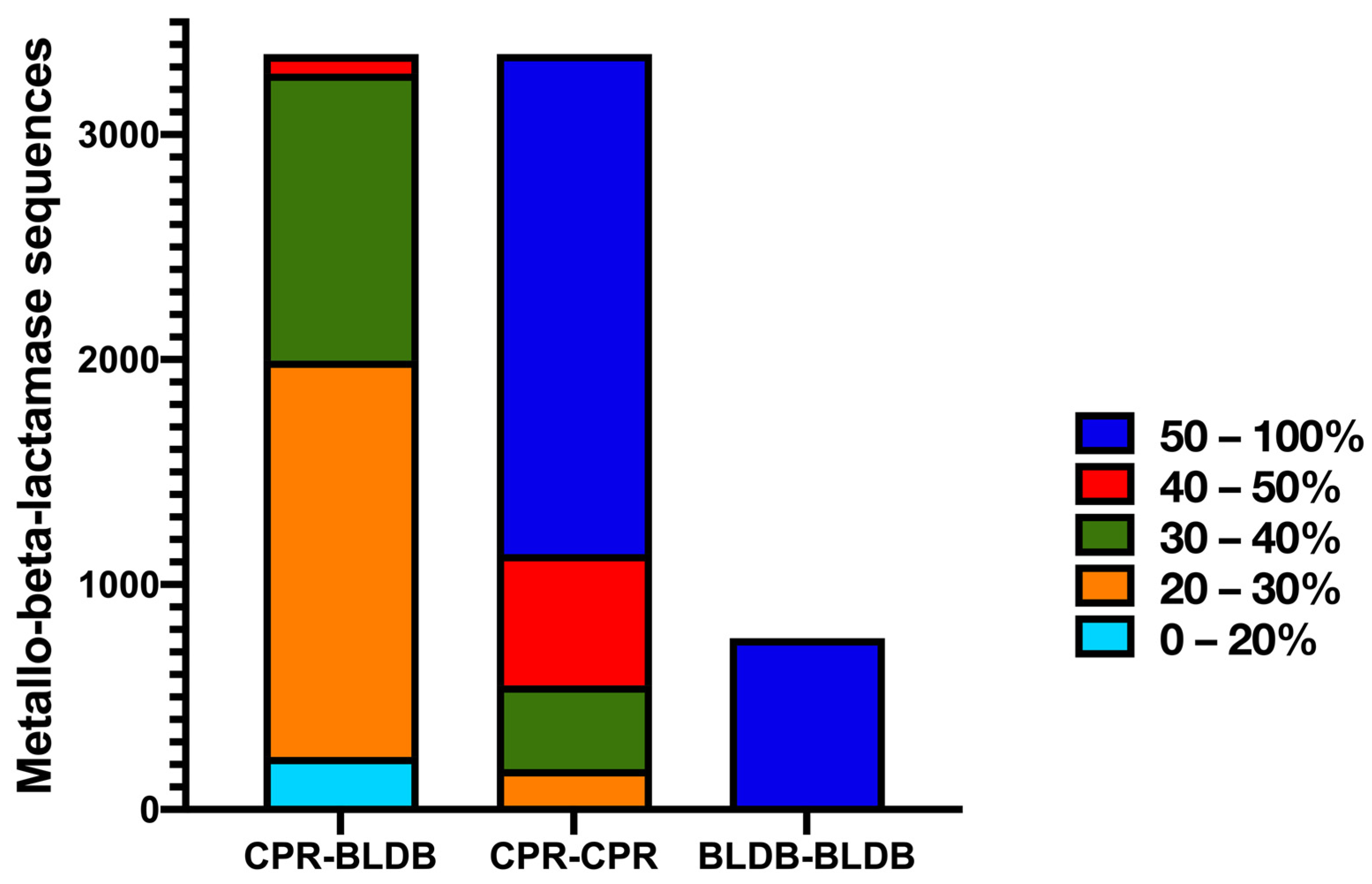{kind=link}
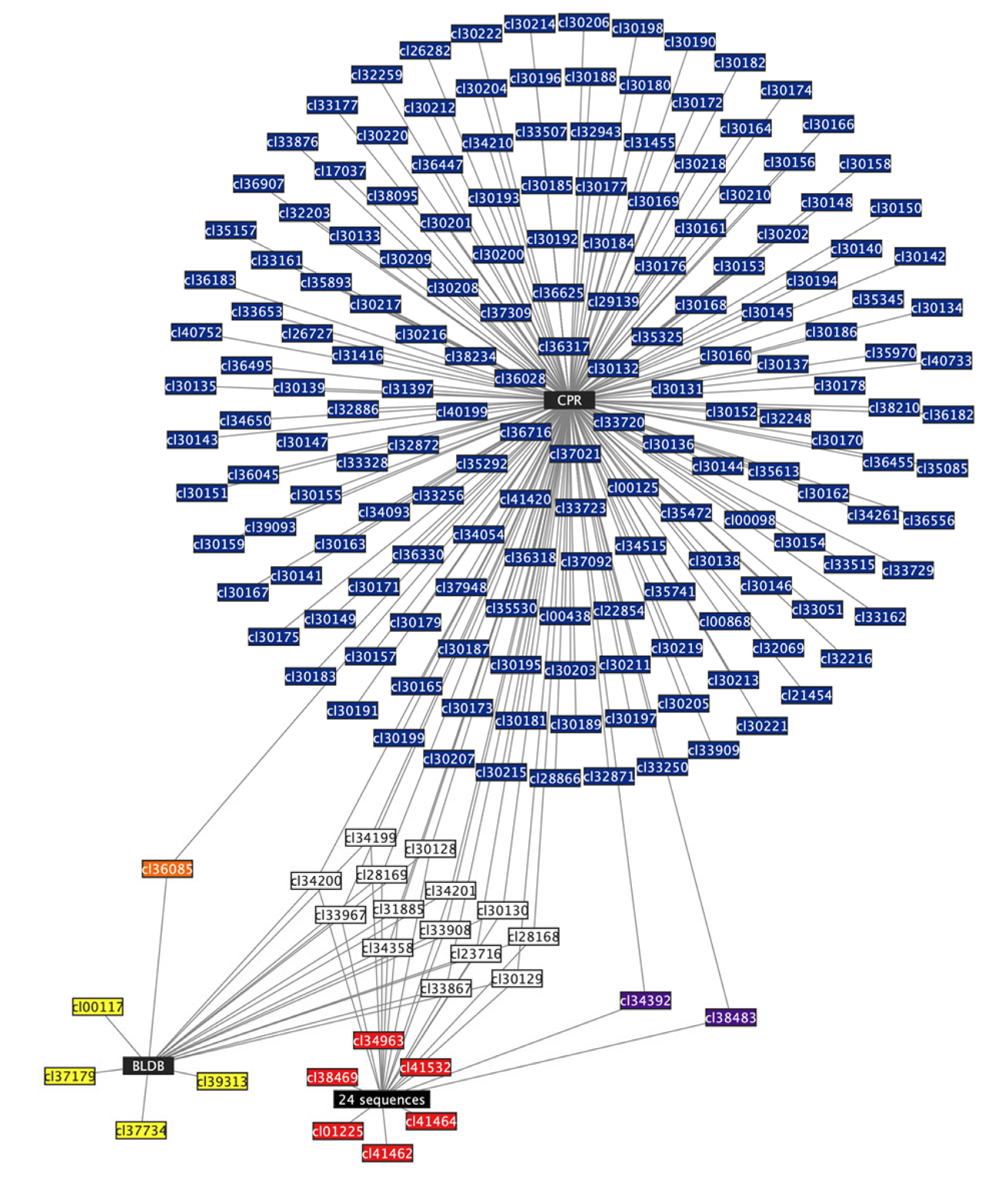{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. CPR MBL-like Sequences: Different Sizes and Divergent Sequences
2.2. The CPR MBL-like Sequences Are Rich in Various Protein Functions
2.3. CPR MBL-like Sequences Have Different Protein Profiles and Many Potential Functions
3. Discussion
4. Materials and Methods
4.1. CPR MBL-like Sequences
4.2. Data Set Curation for Protein Function Profiling
4.3. Construction of the Sequence Similarity Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parks, D.H. Recovery of Nearly 8000 Metagenome-Assembled Genomes Substantially Expands the Tree of Life. Nat. Microbiol. 2017, 2, 10. [Google Scholar] [CrossRef]
- Kantor, R.S.; Wrighton, K.C.; Handley, K.M.; Sharon, I.; Hug, L.A.; Castelle, C.J.; Thomas, B.C.; Banfield, J.F. Small Genomes and Sparse Metabolisms of Sediment-Associated Bacteria from Four Candidate Phyla. mBio 2013, 4, e00708-13. [Google Scholar] [CrossRef]
- Luef, B.; Frischkorn, K.R.; Wrighton, K.C.; Holman, H.-Y.N.; Birarda, G.; Thomas, B.C.; Singh, A.; Williams, K.H.; Siegerist, C.E.; Tringe, S.G.; et al. Diverse Uncultivated Ultra-Small Bacterial Cells in Groundwater. Nat. Commun. 2015, 6, 6372. [Google Scholar] [CrossRef]
- Wrighton, K.C.; Thomas, B.C.; Sharon, I.; Miller, C.S.; Castelle, C.J.; VerBerkmoes, N.C.; Wilkins, M.J.; Hettich, R.L.; Lipton, M.S.; Williams, K.H.; et al. Fermentation, Hydrogen, and Sulfur Metabolism in Multiple Uncultivated Bacterial Phyla. Science 2012, 337, 1661–1665. [Google Scholar] [CrossRef]
- Bernard, C.; Lannes, R.; Li, Y.; Bapteste, É.; Lopez, P. Rich Repertoire of Quorum Sensing Protein Coding Sequences in CPR and DPANN Associated with Interspecies and Interkingdom Communication. mSystems 2020, 5, 10–1128. [Google Scholar] [CrossRef]
- Bedree, J.K.; Bor, B.; Cen, L.; Edlund, A.; Lux, R.; McLean, J.S.; Shi, W.; He, X. Quorum Sensing Modulates the Epibiotic-Parasitic Relationship Between Actinomyces Odontolyticus and Its Saccharibacteria Epibiont, a Nanosynbacter Lyticus Strain, TM7x. Front. Microbiol. 2018, 9, 2049. [Google Scholar] [CrossRef]
- Méheust, R.; Burstein, D.; Castelle, C.J.; Banfield, J.F. The Distinction of CPR Bacteria from Other Bacteria Based on Protein Family Content. Nat. Commun. 2019, 10, 4173. [Google Scholar] [CrossRef]
- Orsi, W.D.; Richards, T.A.; Francis, W.R. Predicted Microbial Secretomes and Their Target Substrates in Marine Sediment. Nat. Microbiol. 2018, 3, 32–37. [Google Scholar] [CrossRef]
- Anantharaman, K.; Brown, C.T.; Hug, L.A.; Sharon, I.; Castelle, C.J.; Probst, A.J.; Thomas, B.C.; Singh, A.; Wilkins, M.J.; Karaoz, U.; et al. Thousands of Microbial Genomes Shed Light on Interconnected Biogeochemical Processes in an Aquifer System. Nat. Commun. 2016, 7, 13219. [Google Scholar] [CrossRef]
- Starr, E.P.; Shi, S.; Blazewicz, S.J.; Probst, A.J.; Herman, D.J.; Firestone, M.K.; Banfield, J.F. Stable Isotope Informed Genome-Resolved Metagenomics Reveals That Saccharibacteria Utilize Microbially-Processed Plant-Derived Carbon. Microbiome 2018, 6, 122. [Google Scholar] [CrossRef]
- Castelle, C.J.; Brown, C.T.; Anantharaman, K.; Probst, A.J.; Huang, R.H.; Banfield, J.F. Biosynthetic Capacity, Metabolic Variety and Unusual Biology in the CPR and DPANN Radiations. Nat. Rev. Microbiol. 2018, 16, 629–645. [Google Scholar] [CrossRef]
- Hugenholtz, P.; Tyson, G.W.; Webb, R.I.; Wagner, A.M.; Blackall, L.L. Investigation of Candidate Division TM7, a Recently Recognized Major Lineage of the Domain Bacteria with No Known Pure-Culture Representatives. Appl. Environ. Microbiol. 2001, 67, 411–419. [Google Scholar] [CrossRef]
- Danczak, R.E.; Johnston, M.D.; Kenah, C.; Slattery, M.; Wrighton, K.C.; Wilkins, M.J. Members of the Candidate Phyla Radiation Are Functionally Differentiated by Carbon- and Nitrogen-Cycling Capabilities. Microbiome 2017, 5, 112. [Google Scholar] [CrossRef]
- Naud, S.; Ibrahim, A.; Valles, C.; Maatouk, M.; Bittar, F.; Tidjani Alou, M.; Raoult, D. Candidate Phyla Radiation, an Underappreciated Division of the Human Microbiome, and Its Impact on Health and Disease. Clin. Microbiol. Rev. 2022, 35, e0014021. [Google Scholar] [CrossRef]
- Tan, S.H.; Normi, Y.M.; Leow, A.T.C.; Salleh, A.B.; Murad, A.M.A.; Mahadi, N.M.; Rahman, M.B.A. Danger Lurking in the “Unknowns”: Structure-to-Function Studies of Hypothetical Protein Bleg1_2437 from Bacillus Lehensis G1 Alkaliphile Revealed an Evolutionary Divergent B3 Metallo-Beta-Lactamase. J. Biochem. 2017, 161, 167–186. [Google Scholar] [CrossRef]
- Maatouk, M.; Ibrahim, A.; Rolain, J.-M.; Merhej, V.; Bittar, F. Small and Equipped: The Rich Repertoire of Antibiotic Resistance Genes in Candidate Phyla Radiation Genomes. Msystems 2021, 6, e00898-21. [Google Scholar] [CrossRef]
- Allewell, N.M. Thematic Minireview Series on Enzyme Evolution in the Post-Genomic Era. J. Biol. Chem. 2012, 287, 1–2. [Google Scholar] [CrossRef]
- Schnoes, A.M.; Brown, S.D.; Dodevski, I.; Babbitt, P.C. Annotation Error in Public Databases: Misannotation of Molecular Function in Enzyme Superfamilies. PLoS Comput. Biol. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Brown, S.D.; Babbitt, P.C. New Insights about Enzyme Evolution from Large Scale Studies of Sequence and Structure Relationships. J. Biol. Chem. 2014, 289, 30221–30228. [Google Scholar] [CrossRef]
- Meroueh, S.O.; Minasov, G.; Lee, W.; Shoichet, B.K.; Mobashery, S. Structural Aspects for Evolution of β-Lactamases from Penicillin-Binding Proteins. J. Am. Chem. Soc. 2003, 125, 9612–9618. [Google Scholar] [CrossRef]
- Pabis, A.; Risso, V.A.; Sanchez-Ruiz, J.M.; Kamerlin, S.C. Cooperativity and Flexibility in Enzyme Evolution. Curr. Opin. Struct. Biol. 2018, 48, 83–92. [Google Scholar] [CrossRef]
- Meini, M.-R.; Tomatis, P.E.; Weinreich, D.M.; Vila, A.J. Quantitative Description of a Protein Fitness Landscape Based on Molecular Features. Mol. Biol. Evol. 2015, 32, 1774–1787. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef]
- Ambler, R.P.; Coulson, A.F.; Frère, J.M.; Ghuysen, J.M.; Joris, B.; Forsman, M.; Levesque, R.C.; Tiraby, G.; Waley, S.G. A Standard Numbering Scheme for the Class A Beta-Lactamases. Biochem. J. 1991, 276, 269–270. [Google Scholar] [CrossRef]
- Bebrone, C. Metallo-Beta-Lactamases (Classification, Activity, Genetic Organization, Structure, Zinc Coordination) and Their Superfamily. Biochem. Pharmacol. 2007, 74, 1686–1701. [Google Scholar] [CrossRef]
- Galán, J.-C.; González-Candelas, F.; Rolain, J.-M.; Cantón, R. Antibiotics as Selectors and Accelerators of Diversity in the Mechanisms of Resistance: From the Resistome to Genetic Plasticity in the β-Lactamases World. Front. Microbiol. 2013, 4, 9. [Google Scholar] [CrossRef]
- Bahr, G.; González, L.J.; Vila, A.J. Metallo-β-Lactamases in the Age of Multidrug Resistance: From Structure and Mechanism to Evolution, Dissemination, and Inhibitor Design. Chem. Rev. 2021, 121, 7957–8094. [Google Scholar] [CrossRef]
- Baier, F.; Tokuriki, N. Connectivity between Catalytic Landscapes of the Metallo-β-Lactamase Superfamily. J. Mol. Biol. 2014, 426, 2442–2456. [Google Scholar] [CrossRef]
- Keshri, V.; Chabrière, E.; Pinault, L.; Colson, P.; Diene, S.M.; Rolain, J.-M.; Raoult, D.; Pontarotti, P. Promiscuous Enzyme Activity as a Driver of Allo and Iso Convergent Evolution, Lessons from the β-Lactamases. Int. J. Mol. Sci. 2020, 21, 6260. [Google Scholar] [CrossRef]
- O’Brien, P.J.; Herschlag, D. Catalytic Promiscuity and the Evolution of New Enzymatic Activities. Chem. Biol. 1999, 6, R91–R105. [Google Scholar] [CrossRef]
- Atkinson, H.J.; Morris, J.H.; Ferrin, T.E.; Babbitt, P.C. Using Sequence Similarity Networks for Visualization of Relationships Across Diverse Protein Superfamilies. PLoS ONE 2009, 4, e4345. [Google Scholar] [CrossRef]
- Jensen, R.A. Enzyme Recruitment in Evolution of New Function. Annu. Rev. Microbiol. 1976, 30, 409–425. [Google Scholar] [CrossRef]
- Diene, S.M.; Pinault, L.; Armstrong, N.; Azza, S.; Keshri, V.; Khelaifia, S.; Chabrière, E.; Caetano-Anolles, G.; Rolain, J.-M.; Pontarotti, P.; et al. Dual RNase and β-Lactamase Activity of a Single Enzyme Encoded in Archaea. Life 2020, 10, 280. [Google Scholar] [CrossRef]
- Mahdavi, S.; Sadeghi, M.; Shokri, R.; Sadegh, B. The Role of Bacteriophages as Important Reservoirs of Extended-Spectrum Beta-Lactamase Genes in Azerbaijan Hospitals. Microb. Drug Resist. 2022, 28, 436–443. [Google Scholar] [CrossRef]
- Colson, P.; Pinault, L.; Azza, S.; Armstrong, N.; Chabriere, E.; La Scola, B.; Pontarotti, P.; Raoult, D. A Protein of the Metallo-Hydrolase/Oxidoreductase Superfamily with Both Beta-Lactamase and Ribonuclease Activity Is Linked with Translation in Giant Viruses. Sci. Rep. 2020, 10, 21685. [Google Scholar] [CrossRef]
- Diene, S.M.; Pinault, L.; Keshri, V.; Armstrong, N.; Khelaifia, S.; Chabrière, E.; Caetano-Anolles, G.; Colson, P.; La Scola, B.; Rolain, J.-M.; et al. Human Metallo-β-Lactamase Enzymes Degrade Penicillin. Sci. Rep. 2019, 9, 12173. [Google Scholar] [CrossRef]
- Hall, B.G.; Barlow, M. Evolution of the Serine Beta-Lactamases: Past, Present and Future. Drug Resist. Updat. 2004, 7, 111–123. [Google Scholar] [CrossRef]
- Daiyasu, H.; Osaka, K.; Ishino, Y.; Toh, H. Expansion of the Zinc Metallo-Hydrolase Family of the Beta-Lactamase Fold. FEBS Lett. 2001, 503, 1–6. [Google Scholar] [CrossRef]
- Diene, S.M.; Pontarotti, P.; Azza, S.; Armstrong, N.; Pinault, L.; Chabrière, E.; Colson, P.; Rolain, J.-M.; Raoult, D. Origin, Diversity, and Multiple Roles of Enzymes with Metallo-β-Lactamase Fold from Different Organisms. Cells 2023, 12, 1752. [Google Scholar] [CrossRef]
- Garau, J. Beta-Lactamases: Current Situation and Clinical Importance. Intensive Care Med. 1994, 20, S5–S9. [Google Scholar] [CrossRef]
- Jacoby, G.A. β-Lactamase Nomenclature. Antimicrob. Agents Chemother. 2006, 50, 1123–1129. [Google Scholar] [CrossRef]
- Resurrecting Ancient Genes: Experimental Analysis of Extinct Molecules|Nature Reviews Genetics. Available online: https://www.nature.com/articles/nrg1324 (accessed on 18 July 2023).
- Risso, V.A.; Gavira, J.A.; Mejia-Carmona, D.F.; Gaucher, E.A.; Sanchez-Ruiz, J.M. Hyperstability and Substrate Promiscuity in Laboratory Resurrections of Precambrian β-Lactamases. Available online: https://pubs.acs.org/doi/pdf/10.1021/ja311630a (accessed on 18 July 2023).
- Maatouk, M.; Ibrahim, A.; Pinault, L.; Armstrong, N.; Azza, S.; Rolain, J.-M.; Bittar, F.; Raoult, D. New Beta-Lactamases in Candidate Phyla Radiation: Owning Pleiotropic Enzymes Is a Smart Paradigm for Microorganisms with a Reduced Genome. Int. J. Mol. Sci. 2022, 23, 5446. [Google Scholar] [CrossRef]
- Gilks, W.R.; Audit, B.; De Angelis, D.; Tsoka, S.; Ouzounis, C.A. Modeling the Percolation of Annotation Errors in a Database of Protein Sequences. Bioinformatics 2002, 18, 1641–1649. [Google Scholar] [CrossRef]
- Gilks, W.R.; Audit, B.; de Angelis, D.; Tsoka, S.; Ouzounis, C.A. Percolation of Annotation Errors through Hierarchically Structured Protein Sequence Databases. Math. Biosci. 2005, 193, 223–234. [Google Scholar] [CrossRef]
- Abriata, L.A.; Salverda, M.L.M.; Tomatis, P.E. Sequence–Function–Stability Relationships in Proteins from Datasets of Functionally Annotated Variants: The Case of TEM β-Lactamases. FEBS Lett. 2012, 586, 3330–3335. [Google Scholar] [CrossRef]
- Pegg, S.C.H.; Brown, S.; Ojha, S.; Huang, C.C.; Ferrin, T.E.; Babbitt, P.C. Representing Structure-Function Relationships in Mechanistically Diverse Enzyme Superfamilies. Pac. Symp. Biocomput. 2005, 2005, 358–369. [Google Scholar]
- Aravind, L. An Evolutionary Classification of the Metallo-Beta-Lactamase Fold Proteins. Silico Biol 1999, 1, 69–91. [Google Scholar]
- Garau, G.; Di Guilmi, A.M.; Hall, B.G. Structure-Based Phylogeny of the Metallo-β-Lactamases. Antimicrob. Agents Chemother. 2005, 49, 2778–2784. [Google Scholar] [CrossRef]
- Mir-Montazeri, B.; Ammelburg, M.; Forouzan, D.; Lupas, A.N.; Hartmann, M.D. Crystal Structure of a Dimeric Archaeal Cleavage and Polyadenylation Specificity Factor. J. Struct. Biol. 2011, 173, 191–195. [Google Scholar] [CrossRef]
- D’Costa, V.M.; Griffiths, E.; Wright, G.D. Expanding the Soil Antibiotic Resistome: Exploring Environmental Diversity. Curr. Opin. Microbiol. 2007, 10, 481–489. [Google Scholar] [CrossRef]
- Kelly, J.A.; Moews, P.C.; Knox, J.R.; Frère, J.M.; Ghuysen, J.M. Penicillin Target Enzyme and the Antibiotic Binding Site. Science 1982, 218, 479–481. [Google Scholar] [CrossRef]
- Bashton, M.; Chothia, C. The Generation of New Protein Functions by the Combination of Domains. Structure 2007, 15, 85–99. [Google Scholar] [CrossRef]
- Aminov, R.I. The Role of Antibiotics and Antibiotic Resistance in Nature. Environ. Microbiol. 2009, 11, 2970–2988. [Google Scholar] [CrossRef]
- O’Maille, P.E.; Malone, A.; Dellas, N.; Hess, B.A.; Smentek, L.; Sheehan, I.; Greenhagen, B.T.; Chappell, J.; Manning, G.; Noel, J.P. Quantitative Exploration of the Catalytic Landscape Separating Divergent Plant Sesquiterpene Synthases. Nat. Chem. Biol. 2008, 4, 617–623. [Google Scholar] [CrossRef]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A New View of the Tree of Life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef]
- Ibrahim, A.; Colson, P.; Merhej, V.; Zgheib, R.; Maatouk, M.; Naud, S.; Bittar, F.; Raoult, D. Rhizomal Reclassification of Living Organisms. Int. J. Mol. Sci. 2021, 22, 5643. [Google Scholar] [CrossRef]
- Czekalski, N.; Sigdel, R.; Birtel, J.; Matthews, B.; Bürgmann, H. Does Human Activity Impact the Natural Antibiotic Resistance Background? Abundance of Antibiotic Resistance Genes in 21 Swiss Lakes. Environ. Int. 2015, 81, 45–55. [Google Scholar] [CrossRef]
- Fondi, M.; Karkman, A.; Tamminen, M.V.; Bosi, E.; Virta, M.; Fani, R.; Alm, E.; McInerney, J.O. “Every Gene Is Everywhere but the Environment Selects”: Global Geolocalization of Gene Sharing in Environmental Samples through Network Analysis. Genome Biol. Evol. 2016, 8, 1388–1400. [Google Scholar] [CrossRef]
- Bhullar, K.; Waglechner, N.; Pawlowski, A.; Koteva, K.; Banks, E.D.; Johnston, M.D.; Barton, H.A.; Wright, G.D. Antibiotic Resistance Is Prevalent in an Isolated Cave Microbiome. PLoS ONE 2012, 7, e34953. [Google Scholar] [CrossRef]
- Cristóbal-Azkarate, J.; Dunn, J.C.; Day, J.M.W.; Amábile-Cuevas, C.F. Resistance to Antibiotics of Clinical Relevance in the Fecal Microbiota of Mexican Wildlife. PLoS ONE 2014, 9, e107719. [Google Scholar] [CrossRef]
- Nesme, J.; Cécillon, S.; Delmont, T.O.; Monier, J.-M.; Vogel, T.M.; Simonet, P. Large-Scale Metagenomic-Based Study of Antibiotic Resistance in the Environment. Curr. Biol. 2014, 24, 1096–1100. [Google Scholar] [CrossRef]
- Woappi, Y.; Gabani, P.; Singh, A.; Singh, O.V. Antibiotrophs: The Complexity of Antibiotic-Subsisting and Antibiotic-Resistant Microorganisms. Crit. Rev. Microbiol. 2016, 42, 17–30. [Google Scholar] [CrossRef]
- Crofts, T.S.; Wang, B.; Spivak, A.; Gianoulis, T.A.; Forsberg, K.J.; Gibson, M.K.; Johnsky, L.A.; Broomall, S.M.; Rosenzweig, C.N.; Skowronski, E.W.; et al. Shared Strategies for β-Lactam Catabolism in the Soil Microbiome. Nat. Chem. Biol. 2018, 14, 556–564. [Google Scholar] [CrossRef]
- Singh, R.; Saxena, A.; Singh, H. Identification of Group Specific Motifs in Beta-Lactamase Family of Proteins. J. Biomed. Sci. 2009, 16, 109. [Google Scholar] [CrossRef]
- PHYRE2 Protein Fold Recognition Server. Available online: http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index (accessed on 4 May 2020).
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Naas, T.; Oueslati, S.; Bonnin, R.A.; Dabos, M.L.; Zavala, A.; Dortet, L.; Retailleau, P.; Iorga, B.I. Beta-Lactamase Database (BLDB)—Structure and Function. J. Enzym. Inhib. Med. Chem. 2017, 32, 917–919. [Google Scholar] [CrossRef]
- Ibrahim, A.; Maatouk, M.; Rajaonison, A.; Zgheib, R.; Haddad, G.; Khalil, J.B.; Raoult, D.; Bittar, F. Adapted Protocol for Saccharibacteria Cocultivation: Two New Members Join the Club of Candidate Phyla Radiation. Microbiol. Spectr. 2021, 9, e01069-21. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maatouk, M.; Merhej, V.; Pontarotti, P.; Ibrahim, A.; Rolain, J.-M.; Bittar, F. Metallo-Beta-Lactamase-like Encoding Genes in Candidate Phyla Radiation: Widespread and Highly Divergent Proteins with Potential Multifunctionality. Microorganisms 2023, 11, 1933. https://doi.org/10.3390/microorganisms11081933
Maatouk M, Merhej V, Pontarotti P, Ibrahim A, Rolain J-M, Bittar F. Metallo-Beta-Lactamase-like Encoding Genes in Candidate Phyla Radiation: Widespread and Highly Divergent Proteins with Potential Multifunctionality. Microorganisms. 2023; 11(8):1933. https://doi.org/10.3390/microorganisms11081933
Chicago/Turabian StyleMaatouk, Mohamad, Vicky Merhej, Pierre Pontarotti, Ahmad Ibrahim, Jean-Marc Rolain, and Fadi Bittar. 2023. "Metallo-Beta-Lactamase-like Encoding Genes in Candidate Phyla Radiation: Widespread and Highly Divergent Proteins with Potential Multifunctionality" Microorganisms 11, no. 8: 1933. https://doi.org/10.3390/microorganisms11081933
APA StyleMaatouk, M., Merhej, V., Pontarotti, P., Ibrahim, A., Rolain, J.-M., & Bittar, F. (2023). Metallo-Beta-Lactamase-like Encoding Genes in Candidate Phyla Radiation: Widespread and Highly Divergent Proteins with Potential Multifunctionality. Microorganisms, 11(8), 1933. https://doi.org/10.3390/microorganisms11081933