
Faecal Microbiota Characterisation of Potamochoerus porcus Living in a Controlled Environment

 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Bifidobacterial Enumeration, Isolation, Genotyping, and Identification
2.3. DNA Extraction and Sequencing
2.4. Statistical Analysis
3. Results
3.1. Bifidobacterial Enumeration, Isolation, Genotyping and Identification
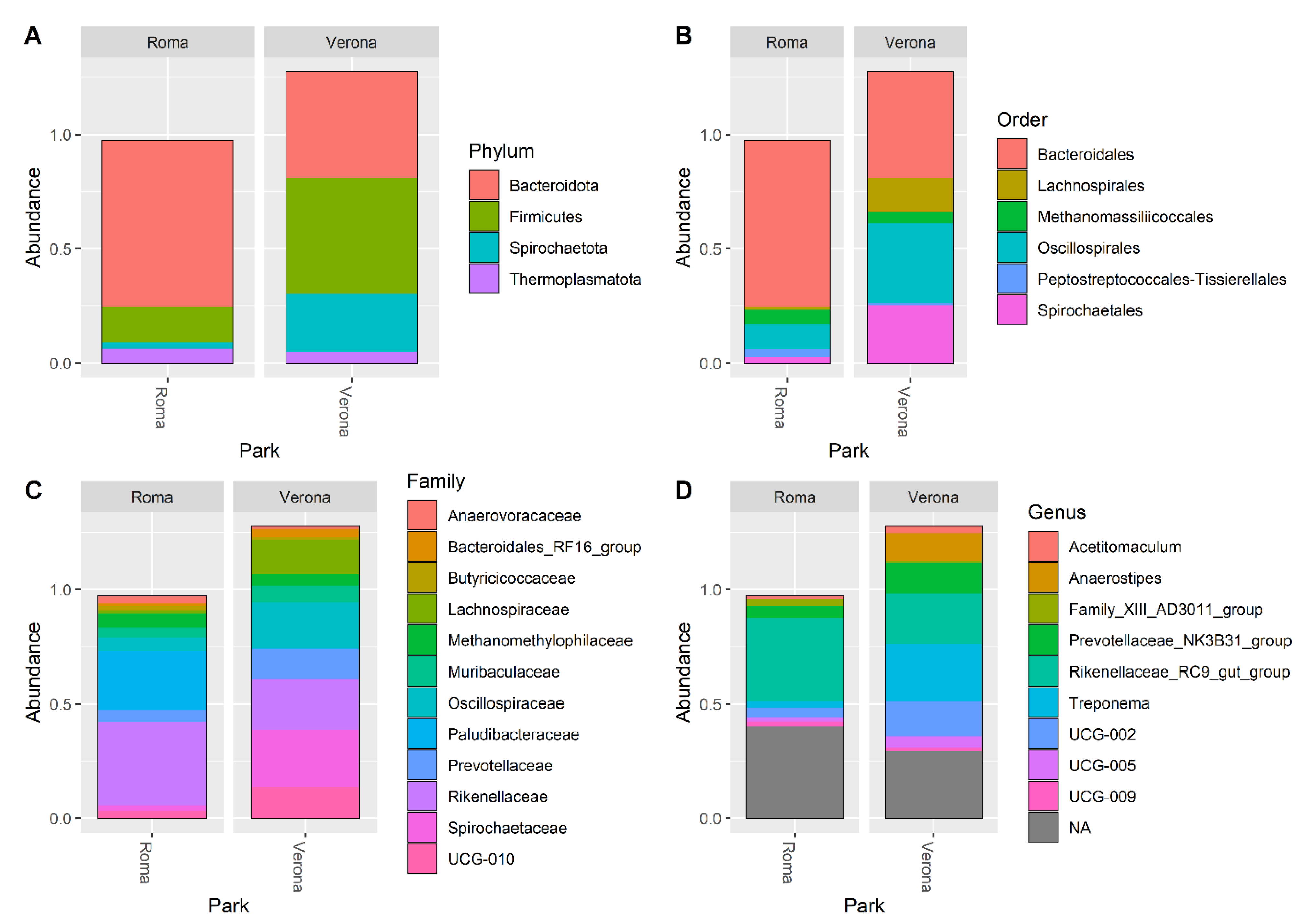
3.2. Microbiota Composition of RRHs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ahn, J.; Hayes, R.B. Environmental Influences on the Human Microbiome and Implications for Noncommunicable Disease. Annu. Rev. Public Health 2021, 42, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, A.; Martin Bideguren, G.; Aizpurua, O. Diversity and compositional changes in the gut microbiota of wild and captive vertebrates: A meta-analysis. Sci. Rep. 2021, 11, 22660. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, G.; van Sinderen, D.; Ventura, M. The genus Bifidobacterium: From genomics to functionality of an important component of the mammalian gut microbiota. Comput. Struct. Biotechnol. J. 2021, 19, 1472–1487. [Google Scholar] [CrossRef]
- Arboleya, S.; Watkins, C.; Stanton, C.; Ross, R.P. Gut Bifidobacteria Populations in Human Health and Aging. Front. Microbiol. 2016, 7, 1204. [Google Scholar] [CrossRef] [PubMed]
- Avershina, E.; Lundgård, K.; Sekelja, M.; Dotterud, C.; Storrø, O.; Øien, T.; Johnsen, R.; Rudi, K. Transition from infant- to adult-like gut microbiota. Environ. Microbiol. 2016, 18, 2226–2236. [Google Scholar] [CrossRef] [PubMed]
- Biavati, B.; Mattarelli, P.; Crociani, F. Identification of Bifidobacteria from Fermented Milk Products. Microbiologica 1992, 15, 7–13. [Google Scholar]
- Bunesova, V.; Vlkova, E.; Rada, V.; Killer, J.; Musilova, S. Bifidobacteria from the gastrointestinal tract of animals: Differences and similarities. Benef. Microbes 2014, 5, 377–388. [Google Scholar] [CrossRef]
- Chao, A.; Chiu, C. (Eds.) Species Richness: Estimation and Comparison. Wiley StatsRef: Statistics Reference Online; Wiley: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Dallas, J.W.; Warne, R.W. Captivity and Animal Microbiomes: Potential Roles of Microbiota for Influencing Animal Conservation. Microb. Ecol. 2023, 85, 820–838. [Google Scholar] [CrossRef]
- Davis, D.J.; Doerr, H.M.; Grzelak, A.K.; Busi, S.B.; Jasarevic, E.; Ericsson, A.C.; Bryda, E.C. Lactobacillus plantarum attenuates anxiety-related behavior and protects against stress-induced dysbiosis in adult zebrafish. Sci. Rep. 2016, 6, 33726. [Google Scholar] [CrossRef]
- de Jonge, N.; Carlsen, B.; Christensen, M.H.; Pertoldi, C.; Nielsen, J.L. The Gut Microbiome of 54 Mammalian Species. Front. Microbiol. 2022, 13, 886252. [Google Scholar] [CrossRef]
- Diaz, J.; Reese, A.T. Possibilities and limits for using the gut microbiome to improve captive animal health. Anim. Microbiome 2021, 3, 89. [Google Scholar] [CrossRef]
- Dixon, P. Computer Program Review VEGAN, a Package of R Functions for Community Ecology. J. Veg. Sci. 2003, 14, 927–930. Available online: http://doi.wiley.com/10.1111/j.1654-1103.2002.tb02049.x (accessed on 1 January 2023). [CrossRef]
- Dowarah, R.; Verma, A.K.; Agarwal, N. The use of Lactobacillus as an alternative of antibiotic growth promoters in pigs: A review. Anim. Nutr. 2017, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Duguma, B.; Eshete, G.; Zewdu, T.; Tolera, A. Comparison of Nutritive Value of Alfalfa, Rhodes Hay, Cynodon Pasture and Linseed Cake–Maize Mixture at Hawassa College of Agriculture, Ethiopia. Acad. J. Nutr. 2014, 3, 19–21. [Google Scholar] [CrossRef]
- Grubb, P. The growth, ecology and population structure of giant tortoises on Aldabra. Philos. Trans. R. Soc. London. B Biol. Sci. 1971, 260, 327–372. [Google Scholar] [CrossRef]
- Guaraldi, F.; Salvatori, G. Effect of breast and formula feeding on gut microbiota shaping in newborns. Front. Cell. Infect. Microbiol. 2012, 2, 94. [Google Scholar] [CrossRef]
- Holman, D.B.; Brunelle, B.W.; Trachsel, J.; Allen, H.K. Meta-analysis to Define a Core Microbiota in the Swine Gut. MSystems 2017, 2, e00004-17. [Google Scholar] [CrossRef]
- Inoue, R.; Tsukahara, T.; Nakanishi, N.; Ushida, K. Development of the intestinal microbiota in the piglet. J. Gen. Appl. Microbiol. 2005, 51, 257–265. [Google Scholar] [CrossRef]
- Inoue, R.; Ushida, K. Vertical and horizontal transmission of intestinal commensal bacteria in the rat model. FEMS Microbiol. Ecol. 2003, 46, 213–219. [Google Scholar] [CrossRef]
- Jin Song, S.; Woodhams, D.C.; Martino, C.; Allaband, C.; Mu, A.; Javorschi-Miller-Montgomery, S.; Suchodolski, J.S.; Knight, R. Engineering the microbiome for animal health and conservation. Exp. Biol. Med. 2019, 244, 494–504. [Google Scholar] [CrossRef]
- Katouli, M.; Lund, A.; Wallgren, P.; Kühn, I.; Söderlind, O.; Möllby, R. Metabolic fingerprinting and fermentative capacity of the intestinal flora of pigs during pre- and post-weaning periods. J. Appl. Microbiol. 1997, 83, 147–154. [Google Scholar] [CrossRef]
- Konstantinov, S.R.; Awati, A.A.; Williams, B.A.; Miller, B.G.; Jones, P.; Stokes, C.R.; Akkermans, A.D.L.; Smidt, H.; De Vos, W.M. Post-natal development of the porcine microbiota composition and activities. Environ. Microbiol. 2006, 8, 1191–1199. [Google Scholar] [CrossRef]
- Kostic, A.D.; Howitt, M.R.; Garrett, W.S. Exploring host-microbiota interactions in animal models and humans. Genes Dev. 2013, 27, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Leslie, D.M.; Huffman, B.A. Potamochoerus porcus (Artiodactyla: Suidae). Mamm. Species 2015, 47, 15–31. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Duranti, S.; Milani, C.; Mancabelli, L.; Turroni, F.; Sinderen, D.v.; Ventura, M. Uncovering Bifidobacteria via Targeted Sequencing of the Mammalian Gut Microbiota. Microorganisms 2019, 7, 535. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ren, W.; Smidt, H.; Wright, A.-D.G.; Yu, B.; Schyns, G.; McCormack, U.M.; Cowieson, A.J.; Yu, J.; He, J.; et al. Dynamic Distribution of Gut Microbiota in Pigs at Different Growth Stages: Composition and Contribution. Microbiol. Spectr. 2022, 10, e0068821. [Google Scholar] [CrossRef]
- Mackie, R.I.; Sghir, A.; Gaskins, H.R. Developmental microbial ecology of the neonatal gastrointestinal tract. Am. J. Clin. Nutr. 1999, 69, 1035S–1045S. [Google Scholar] [CrossRef]
- Magnusson, K.R.; Hauck, L.; Jeffrey, B.M.; Elias, V.; Humphrey, A.; Nath, R.; Perrone, A.; Bermudez, L.E. Relationships between diet-related changes in the gut microbiome and cognitive flexibility. Neuroscience 2015, 300, 128–140. [Google Scholar] [CrossRef]
- Martinez Arbizu, P. Pairwiseadonis: Pairwise Multilevel Comparison Using Adonis; R Package Version 0.4. 2020. Available online: https://github.com/pmartinezarbizu/pairwiseAdonis (accessed on 1 January 2023).
- Matsuki, T.; Watanabe, K.; Fujimoto, J.; Takada, T.; Tanaka, R. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl. Environ. Microbiol. 2004, 70, 7220–7228. [Google Scholar] [CrossRef]
- McKenzie, V.J.; Song, S.J.; Delsuc, F.; Prest, T.L.; Oliverio, A.M.; Korpita, T.M.; Alexiev, A.; Amato, K.R.; Metcalf, J.L.; Kowalewski, M.; et al. The Effects of Captivity on the Mammalian Gut Microbiome. Integr. Comp. Biol. 2017, 57, 690–704. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Michelini, S.; Modesto, M.; Oki, K.; Stenico, V.; Stefanini, I.; Biavati, B.; Watanabe, K.; Ferrara, A.; Mattarelli, P. Isolation and identification of cultivable Bifidobacterium spp. from the faeces of 5 baby common marmosets (Callithrix jacchus L.). Anaerobe 2015, 33, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, L.L.; Bendixen, C.; Jakobsen, M.; Jensen, B.B. Enumeration of Bifidobacteria in Gastrointestinal Samples from Piglets. Appl. Environ. Microbiol. 2003, 69, 654. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Mancabelli, L.; Ferrario, C.; Mangifesta, M.; Hevia, A.; Viappiani, A.; Scholz, M.; et al. Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Sci. Rep. 2015, 5, 15782. [Google Scholar] [CrossRef]
- Milani, C.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Duranti, S.; Viappiani, A.; Mangifesta, M.; Segata, N.; van Sinderen, D.; Ventura, M. Evaluation of bifidobacterial community composition in the human gut by means of a targeted amplicon sequencing (ITS) protocol. FEMS Microbiol. Ecol. 2014, 90, 493–503. [Google Scholar] [CrossRef]
- Modesto, M.; Michelini, S.; Oki, K.; Biavati, B.; Watanabe, K.; Mattarelli, P. Bifidobacterium catulorum sp. nov., a novel taxon from the faeces of the baby common marmoset (Callithrix jacchus). Int. J. Syst. Evol. Microbiol. 2018, 68, 575–581. [Google Scholar] [CrossRef]
- Modesto, M.; Michelini, S.; Sansosti, M.C.M.C.; De Filippo, C.; Cavalieri, D.; Qvirist, L.; Andlid, T.; Spiezio, C.; Sandri, C.; Pascarelli, S.; et al. Bifidobacterium callitrichidarum sp. Nov. from the faeces of the emperor tamarin (saguinus imperator). Int. J. Syst. Evol. Microbiol. 2018, 68, 141–148. [Google Scholar] [CrossRef]
- Modesto, M.; Michelini, S.; Stefanini, I.; Sandri, C.; Spiezio, C.; Pisi, A.; Filippini, G.; Biavati, B.; Mattarelli, P. Bifidobacterium lemurum sp. nov., from faeces of the ring-tailed lemur (Lemur catta). Int. J. Syst. Evol. Microbiol. 2015, 65, 1726–1734. [Google Scholar] [CrossRef]
- Modesto, M.; Satti, M.; Watanabe, K.; Scarafile, D.; Huang, C.H.; Liou, J.S.; Tamura, T.; Saito, S.; Watanabe, M.; Mori, K.; et al. Phylogenetic characterization of two novel species of the genus Bifidobacterium: Bifidobacterium saimiriisciurei sp. nov. and Bifidobacterium platyrrhinorum sp. nov. Syst. Appl. Microbiol. 2020, 43, 126111. [Google Scholar] [CrossRef]
- Perofsky, A.C.; Lewis, R.J.; Meyers, L.A. Terrestriality and bacterial transfer: A comparative study of gut microbiomes in sympatric Malagasy mammals. ISME J. 2019, 13, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Petri, D.; Hill, J.E.; Van Kessel, A.G. Microbial succession in the gastrointestinal tract (GIT) of the preweaned pig. Livest. Sci. 2010, 133, 107–109. [Google Scholar] [CrossRef]
- R Core Team. A Language and Environment for Statistical Computing. In R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2019. [Google Scholar]
- Rabetafika, H.N.; Razafindralambo, A.; Ebenso, B.; Razafindralambo, H.L. Probiotics as Antibiotic Alternatives for Human and Animal Applications. Encyclopedia 2023, 3, 561–581. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Thukral, A.K. A review on measurement of Alpha diversity in biology. Agric. Res. J. 2017, 54, 1–10. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Marked alterations in the distal gut microbiome linked to diet-induced obesity. Cell Host Microbe 2008, 3, 213. [Google Scholar] [CrossRef]
- Turroni, F.; Foroni, E.; Pizzetti, P.; Giubellini, V.; Ribbera, A.; Merusi, P.; Cagnasso, P.; Bizzarri, B.; De’Angelis, G.L.; Shanahan, F.; et al. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Appl. Environ. Microbiol. 2009, 75, 1534–1545. [Google Scholar] [CrossRef]
- Ushida, K.; Kameue, C.; Tsukahara, T.; Fukuta, K.; Nakanishi, N. Decreasing traits of fecal immunoglobulin A in neonatal and weaning piglets. J. Vet. Med. Sci. 2008, 70, 849–852. [Google Scholar] [CrossRef]
- Ushida, K.; Tsuchida, S.; Ogura, Y.; Toyoda, A.; Maruyama, F. Domestication and cereal feeding developed domestic pig-type intestinal microbiota in animals of suidae. Anim. Sci. J. 2015, 87, 835–841. [Google Scholar] [CrossRef]
- Ventura, M.; Turroni, F.; Motherway, M.O.; MacSharry, J.; van Sinderen, D. Host-microbe interactions that facilitate gut colonization by commensal bifidobacteria. Trends Microbiol. 2012, 20, 467–476. [Google Scholar] [CrossRef]
- Yang, G.; Shi, C.; Zhang, S.; Liu, Y.; Li, Z.; Gao, F.; Cui, Y.; Yan, Y.; Li, M. Characterization of the bacterial microbiota composition and evolution at different intestinal tract in wild pigs (Sus scrofa ussuricus). PeerJ 2020, 8, e9124. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Age | Sex | Born at | Living in | Counts |
|---|---|---|---|---|---|
| RRHa | Adult | male | Touroparc (Francia) | Parco Natura Viva, Verona (Italy) | 2 × 106 |
| RRHb | Adult | female | Westfalischer (Germania) | Parco Natura Viva, Verona (Italy) | 5 × 106 |
| RRHc | Juvenile | male | Bussolengo | Parco Natura Viva, Verona (Italy) | 3 × 107 |
| RRHd | Adult | male | Wroclaw (Polonia) | Bioparco, Rome (Italy) | 2 × 106 |
| RRHe | Adult | male | Wroclaw (Polonia) | Bioparco, Rome (Italy) | 2 × 106 |
| Number of Isolates | |||||
|---|---|---|---|---|---|
| Species | Verona | Rome | |||
| RRHa | RRHb | RRHc | RRHd | RRHe | |
| B. thermoacidophilum | 7 | 5 | 6 | 0 | 0 |
| B. boum | 2 | 4 | 2 | 0 | 0 |
| B. porcinum | 0 | 0 | 0 | 11 | 8 |
| Site | baseMean1 | log2FoldChange2 | lfcSE3 | Padj4 | Taxa Name |
|---|---|---|---|---|---|
| Family level | |||||
| Rome | 32.57 | −8.58 | 2.04 | 0.000 | Oscillospirales UCG-011 |
| Rome | 19.98 | −7.87 | 2.32 | 0.005 | Paracaedibacteraceae |
| Rome | 77.48 | −4.30 | 0.79 | 0.000 | Sutterellaceae |
| Rome | 4014.90 | −3.95 | 0.82 | 0.000 | Paludibacteraceae |
| Verona | 6104.15 | 2.02 | 0.59 | 0.005 | Spirochaetaceae |
| Verona | 163.14 | 2.76 | 0.90 | 0.016 | Akkermansiaceae |
| Verona | 43.72 | 3.32 | 0.95 | 0.005 | Defluviitaleaceae |
| Verona | 294.27 | 7.19 | 2.62 | 0.039 | Bacteroidales_p-2534-18B5_gut_group |
| Verona | 210.07 | 11.07 | 1.68 | 0.000 | Lactobacillaceae |
| Verona | 21.74 | 20.63 | 4.41 | 0.000 | Corynebacteriaceae |
| Genus level | |||||
| Rome | 17.53 | −7.98 | 2.28 | 0.01 | Lachnospiraceae_UCG-003 |
| Rome | 10.96 | −7.30 | 2.66 | 0.04 | Cellulosilyticum |
| Rome | 59.64 | −5.80 | 1.68 | 0.01 | Prevotellaceae_UCG-004 |
| Rome | 39.19 | −5.18 | 1.84 | 0.04 | Lachnospira |
| Rome | 346.37 | −4.81 | 0.67 | 0.00 | Alloprevotella |
| Rome | 86.85 | −4.73 | 0.91 | 0.00 | Sutterella |
| Verona | 4744.10 | 2.06 | 0.73 | 0.04 | Treponema |
| Verona | 275.20 | 3.36 | 0.97 | 0.01 | Blautia |
| Verona | 249.39 | 3.40 | 0.95 | 0.01 | Candidatus_Soleaferrea |
| Verona | 37.42 | 5.14 | 1.71 | 0.02 | Erysipelotrichaceae_UCG-003 |
| Verona | 1645.90 | 6.34 | 0.94 | 0.00 | Anaerostipes |
| Verona | 11.92 | 6.73 | 2.41 | 0.04 | Dorea |
| Verona | 14.96 | 7.06 | 2.34 | 0.02 | Papillibacter |
| Verona | 67.53 | 9.23 | 1.78 | 0.00 | Lachnospiraceae_XPB1014_group |
| Verona | 174.23 | 10.60 | 1.72 | 0.00 | Lactobacillus |
| Verona | 239.91 | 11.06 | 1.74 | 0.00 | Rikenellaceae_dgA-11_gut_group |
| Verona | 20.93 | 20.36 | 3.63 | 0.00 | Corynebacterium |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarafile, D.; Luise, D.; Motta, V.; Spiezio, C.; Modesto, M.; Porcu, M.M.; Yitzhak, Y.; Correa, F.; Sandri, C.; Trevisi, P.; et al. Faecal Microbiota Characterisation of Potamochoerus porcus Living in a Controlled Environment. Microorganisms 2023, 11, 1542. https://doi.org/10.3390/microorganisms11061542
Scarafile D, Luise D, Motta V, Spiezio C, Modesto M, Porcu MM, Yitzhak Y, Correa F, Sandri C, Trevisi P, et al. Faecal Microbiota Characterisation of Potamochoerus porcus Living in a Controlled Environment. Microorganisms. 2023; 11(6):1542. https://doi.org/10.3390/microorganisms11061542
Chicago/Turabian StyleScarafile, Donatella, Diana Luise, Vincenzo Motta, Caterina Spiezio, Monica Modesto, Marzia Mattia Porcu, Yadid Yitzhak, Federico Correa, Camillo Sandri, Paolo Trevisi, and et al. 2023. "Faecal Microbiota Characterisation of Potamochoerus porcus Living in a Controlled Environment" Microorganisms 11, no. 6: 1542. https://doi.org/10.3390/microorganisms11061542
APA StyleScarafile, D., Luise, D., Motta, V., Spiezio, C., Modesto, M., Porcu, M. M., Yitzhak, Y., Correa, F., Sandri, C., Trevisi, P., & Mattarelli, P. (2023). Faecal Microbiota Characterisation of Potamochoerus porcus Living in a Controlled Environment. Microorganisms, 11(6), 1542. https://doi.org/10.3390/microorganisms11061542