
A First Insight into the Microbial and Viral Communities of Comau Fjord—A Unique Human-Impacted Ecosystem in Patagonia (42∘ S)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sequencing
2.2. Water Nutrients, Elemental Composition, and Physicochemical Analyses
2.3. Distance-Based Analysis and Comparison with Global Ocean Data
2.4. Taxonomic Composition Metagenomes and Viromes
2.5. Metagenomes Functional Potential and Presence of AR Genes
2.6. Viromes Protein Clusters
2.7. Analysis and Visualization
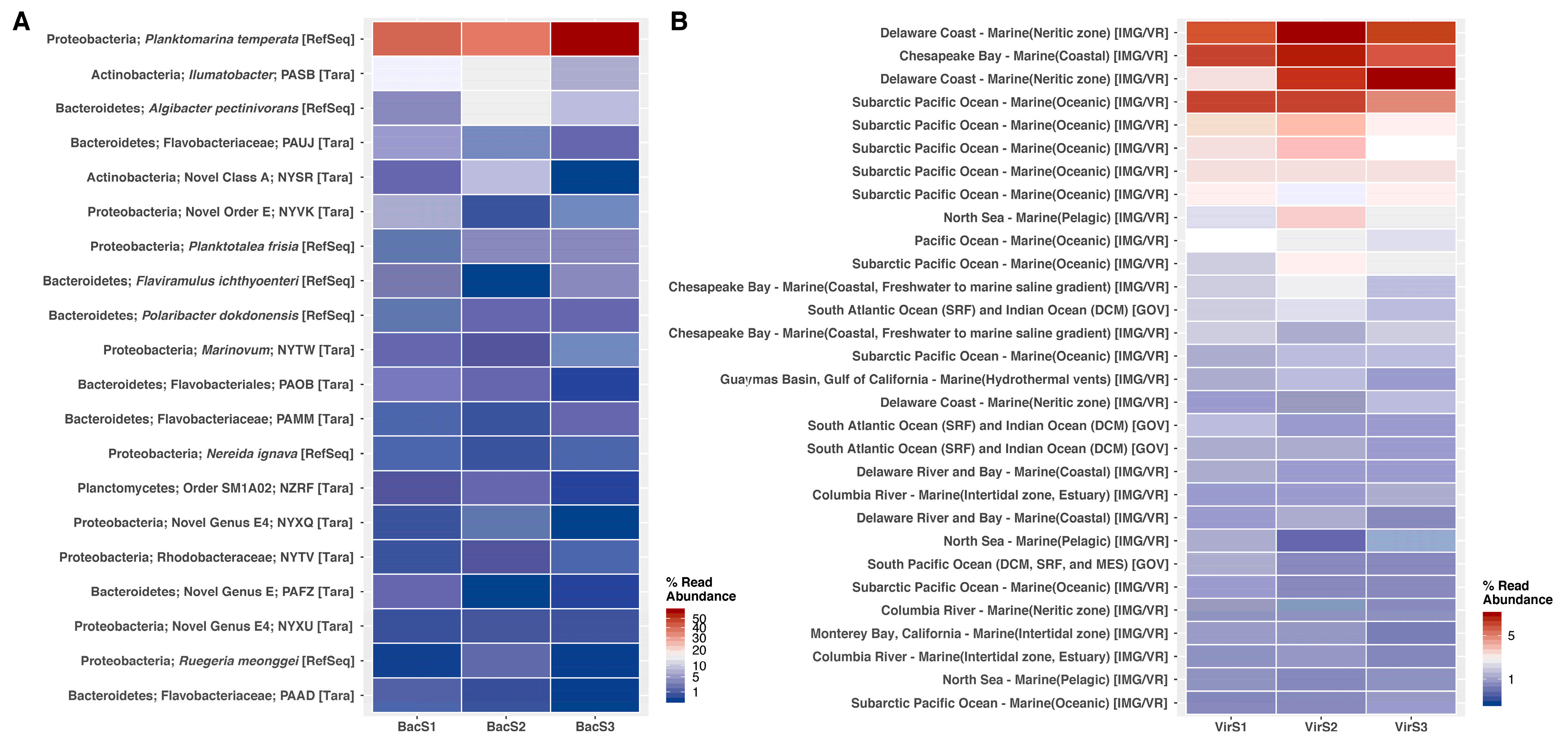
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, Y.-G.; Zhao, Y.; Li, B.; Huang, C.-L.; Zhang, S.-Y.; Yu, S.; Chen, Y.-S.; Zhang, T.; Gillings, M.R.; Su, J.-Q. Continental-scale pollution of estuaries with antibiotic resistance genes. Nat. Microbiol. 2017, 2, 16270. [Google Scholar] [CrossRef]
- Hitchcock, J.N.; Mitrovic, S.M. Microplastic pollution in estuaries across a gradient of human impact. Environ. Pollut. 2019, 247, 457–466. [Google Scholar] [CrossRef]
- Barletta, M.; Lima, A.R.; Costa, M.F. Distribution, sources and consequences of nutrients, persistent organic pollutants, metals and microplastics in South American estuaries. Sci. Total Environ. 2019, 651, 1199–1218. [Google Scholar] [CrossRef]
- Solís, R.; Toro, R.; Gomez, L.; Vélez-Pereira, A.M.; López, M.; Fleming, Z.L.; Fierro, N.; Leiva, M. Long-term airborne particle pollution assessment in the city of Coyhaique, Patagonia, Chile. Urban Clim. 2022, 43, 101144. [Google Scholar] [CrossRef]
- Miglioranza, K.S.; Ondarza, P.M.; Costa, P.G.; De Azevedo, A.; Gonzalez, M.; Shimabukuro, V.M.; Grondona, S.I.; Mitton, F.M.; Barra, R.O.; Wania, F. Spatial and temporal distribution of Persistent Organic Pollutants and current use pesticides in the atmosphere of Argentinean Patagonia. Chemosphere 2021, 266, 129015. [Google Scholar] [CrossRef]
- Galbán-Malagón, C.; Berrojalbiz, N.; Ojeda, M.-J.; Dachs, J. The oceanic biological pump modulates the atmospheric transport of persistent organic pollutants to the Arctic. Nat. Commun. 2012, 3, 862. [Google Scholar] [CrossRef]
- Galbán-Malagón, C.; Cabrerizo, A.; Caballero, G.; Dachs, J. Atmospheric occurrence and deposition of hexachlorobenzene and hexachlorocyclohexanes in the Southern Ocean and Antarctic Peninsula. Atmos. Environ. 2013, 80, 41–49. [Google Scholar] [CrossRef]
- Birch, G.; Cox, B.; Besley, C. The relationship between metal concentrations in seagrass (Zostera capricorni) tissue and ambient fine sediment in modified and near-pristine estuaries (Sydney estuaries, Australia). Mar. Pollut. Bull. 2018, 128, 72–81. [Google Scholar] [CrossRef]
- Marcoleta, A.E.; Arros, P.; Varas, M.A.; Costa, J.; Rojas-Salgado, J.; Berríos-Pastén, C.; Tapia-Fuentes, S.; Silva, D.; Fierro, J.; Canales, N. The highly diverse Antarctic Peninsula soil microbiota as a source of novel resistance genes. Sci. Total Environ. 2022, 810, 152003. [Google Scholar] [CrossRef]
- Martínez, M.L.; Intralawan, A.; Vázquez, G.; Pérez-Maqueo, O.; Sutton, P.; Landgrave, R. The coasts of our world: Ecological, economic and social importance. Ecol. Econ. 2007, 63, 254–272. [Google Scholar] [CrossRef]
- Larsson, D.J.; Flach, C.-F. Antibiotic resistance in the environment. Nat. Rev. Microbiol. 2022, 20, 257–269. [Google Scholar] [CrossRef]
- Manaia, C.M. Assessing the risk of antibiotic resistance transmission from the environment to humans: Non-direct proportionality between abundance and risk. Trends Microbiol. 2017, 25, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.P.; Murray, R.; Scott, A.; Tien, Y.-C.; Lau, C.H.-F.; Tai, V.; Topp, E. Responses of the soil bacterial community, resistome, and mobilome to a decade of annual exposure to macrolide antibiotics. Appl. Environ. Microbiol. 2022, 88, e00316-22. [Google Scholar] [CrossRef] [PubMed]
- Karaolia, P.; Vasileiadis, S.; Michael, S.G.; Karpouzas, D.G.; Fatta-Kassinos, D. Shotgun metagenomics assessment of the resistome, mobilome, pathogen dynamics and their ecological control modes in full-scale urban wastewater treatment plants. J. Hazard. Mater. 2021, 418, 126387. [Google Scholar] [CrossRef] [PubMed]
- Danko, D.; Bezdan, D.; Afshin, E.E.; Ahsanuddin, S.; Bhattacharya, C.; Butler, D.J.; Chng, K.R.; Donnellan, D.; Hecht, J.; Jackson, K. A global metagenomic map of urban microbiomes and antimicrobial resistance. Cell 2021, 184, 3376–3393.e17. [Google Scholar] [CrossRef] [PubMed]
- Sukhum, K.V.; Diorio-Toth, L.; Dantas, G. Genomic and metagenomic approaches for predictive surveillance of emerging pathogens and antibiotic resistance. Clin. Pharmacol. Ther. 2019, 106, 512–524. [Google Scholar] [CrossRef]
- Nava, A.R.; Daneshian, L.; Sarma, H. Antibiotic resistant genes in the environment-exploring surveillance methods and sustainable remediation strategies of antibiotics and ARGs. Environ. Res. 2022, 215, 114212. [Google Scholar] [CrossRef]
- Imchen, M.; Kumavath, R.; Barh, D.; Vaz, A.; Góes-Neto, A.; Tiwari, S.; Ghosh, P.; Wattam, A.R.; Azevedo, V. Comparative mangrove metagenome reveals global prevalence of heavy metals and antibiotic resistome across different ecosystems. Sci. Rep. 2018, 8, 11187. [Google Scholar] [CrossRef]
- Wang, J.; Pan, R.; Dong, P.; Liu, S.; Chen, Q.; Borthwick, A.G.; Sun, L.; Xu, N.; Ni, J. Supercarriers of antibiotic resistome in a world’s large river. Microbiome 2022, 10, 111. [Google Scholar] [CrossRef]
- Karkman, A.; Pärnänen, K.; Larsson, D.J. Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nat. Commun. 2019, 10, 80. [Google Scholar] [CrossRef]
- Buelow, E.; Ploy, M.-C.; Dagot, C. Role of pollution on the selection of antibiotic resistance and bacterial pathogens in the environment. Curr. Opin. Microbiol. 2021, 64, 117–124. [Google Scholar] [CrossRef]
- Zhu, Y.-G.; Johnson, T.A.; Su, J.-Q.; Qiao, M.; Guo, G.-X.; Stedtfeld, R.D.; Hashsham, S.A.; Tiedje, J.M. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. USA 2013, 110, 3435–3440. [Google Scholar] [CrossRef]
- Johnson, T.A.; Stedtfeld, R.D.; Wang, Q.; Cole, J.R.; Hashsham, S.A.; Looft, T.; Zhu, Y.-G.; Tiedje, J.M. Clusters of antibiotic resistance genes enriched together stay together in swine agriculture. MBio 2016, 7, e02214-15. [Google Scholar] [CrossRef]
- McKinney, C.W.; Dungan, R.S.; Moore, A.; Leytem, A.B. Occurrence and abundance of antibiotic resistance genes in agricultural soil receiving dairy manure. FEMS Microbiol. Ecol. 2018, 94, fiy010. [Google Scholar] [CrossRef]
- Chng, K.R.; Li, C.; Bertrand, D.; Ng, A.H.Q.; Kwah, J.S.; Low, H.M.; Tong, C.; Natrajan, M.; Zhang, M.H.; Xu, L. Cartography of opportunistic pathogens and antibiotic resistance genes in a tertiary hospital environment. Nat. Med. 2020, 26, 941–951. [Google Scholar] [CrossRef]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, G.; Song, W.; Ye, C.; Lin, H.; Li, Z.; Liu, W. Plastics in the marine environment are reservoirs for antibiotic and metal resistance genes. Environ. Int. 2019, 123, 79–86. [Google Scholar] [CrossRef]
- Gaze, W.H.; Zhang, L.; Abdouslam, N.A.; Hawkey, P.M.; Calvo-Bado, L.; Royle, J.; Brown, H.; Davis, S.; Kay, P.; Boxall, A. Impacts of anthropogenic activity on the ecology of class 1 integrons and integron-associated genes in the environment. ISME J. 2011, 5, 1253–1261. [Google Scholar] [CrossRef]
- Gillings, M.R.; Gaze, W.H.; Pruden, A.; Smalla, K.; Tiedje, J.M.; Zhu, Y.-G. Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 2015, 9, 1269–1279. [Google Scholar] [CrossRef]
- Tan, L.; Li, L.; Ashbolt, N.; Wang, X.; Cui, Y.; Zhu, X.; Xu, Y.; Yang, Y.; Mao, D.; Luo, Y. Arctic antibiotic resistance gene contamination, a result of anthropogenic activities and natural origin. Sci. Total Environ. 2018, 621, 1176–1184. [Google Scholar] [CrossRef]
- Marti, E.; Variatza, E.; Balcazar, J.L. The role of aquatic ecosystems as reservoirs of antibiotic resistance. Trends Microbiol. 2014, 22, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.D.; Godoy, F.A.; Lee, M.R. Current status of the use of antibiotics and the antimicrobial resistance in the Chilean salmon farms. Front. Microbiol. 2018, 9, 1284. [Google Scholar] [CrossRef] [PubMed]
- Higuera-Llantén, S.; Vásquez-Ponce, F.; Barrientos-Espinoza, B.; Mardones, F.O.; Marshall, S.H.; Olivares-Pacheco, J. Extended antibiotic treatment in salmon farms select multiresistant gut bacteria with a high prevalence of antibiotic resistance genes. PLoS ONE 2018, 13, e0203641. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.D.; Zemelman, R. Bacterial resistance to oxytetracycline in Chilean salmon farming. Aquaculture 2002, 212, 31–47. [Google Scholar] [CrossRef]
- Fernández-Alarcón, C.; Miranda, C.; Singer, R.; López, Y.; Rojas, R.; Bello, H.; Domínguez, M.; González-Rocha, G. Detection of the floR gene in a diversity of florfenicol resistant Gram-negative bacilli from freshwater salmon farms in Chile. Zoonoses Public Health 2010, 57, 181–188. [Google Scholar] [CrossRef]
- Sunagawa, S.; Acinas, S.G.; Bork, P.; Bowler, C.; Eveillard, D.; Gorsky, G.; Guidi, L.; Iudicone, D.; Karsenti, E. Tara Oceans: Towards global ocean ecosystems biology. Nat. Rev. Microbiol. 2020, 18, 428–445. [Google Scholar] [CrossRef]
- Alberti, A.; Poulain, J.; Engelen, S.; Labadie, K.; Romac, S.; Ferrera, I.; Albini, G.; Aury, J.-M.; Belser, C.; Bertrand, A.; et al. Viral to metazoan marine plankton nucleotide sequences from the Tara Oceans expedition. Sci. Data 2017, 4, 170093. [Google Scholar] [CrossRef]
- Duarte, C.M. Seafaring in the 21st century: The Malaspina 2010 circumnavigation expedition. Limnol. Oceanogr. Bull. 2015, 24, 11–14. [Google Scholar] [CrossRef]
- Parthasarathy, H.; Hill, E.; MacCallum, C. Global ocean sampling collection. PLoS Biol. 2007, 5, e83. [Google Scholar] [CrossRef]
- Anderson, R.F. GEOTRACES: Accelerating research on the marine biogeochemical cycles of trace elements and their isotopes. Annu. Rev. Mar. Sci. 2020, 12, 49–85. [Google Scholar] [CrossRef]
- Trevathan-Tackett, S.M.; Sherman, C.D.; Huggett, M.J.; Campbell, A.H.; Laverock, B.; Hurtado-McCormick, V.; Seymour, J.R.; Firl, A.; Messer, L.F.; Ainsworth, T.D. A horizon scan of priorities for coastal marine microbiome research. Nat. Ecol. Evol. 2019, 3, 1509–1520. [Google Scholar] [CrossRef]
- SERNAPESCA. Informe Sobre Uso de Antimicrobianos en la Salmonicultura Nacional; Sub-directorate of Aquaculture, Animal Health Department: Valparaíso, Chile, 2020. [Google Scholar]
- Alarcón-Schumacher, T.; Guajardo-Leiva, S.; Antón, J.; Díez, B. Elucidating Viral Communities During a Phytoplankton Bloom on the West Antarctic Peninsula. Front. Microbiol. 2019, 10, 1014. [Google Scholar] [CrossRef]
- Guajardo-Leiva, S.; Santos, F.; Salgado, O.; Regeard, C.; Quillet, L.; Díez, B. Unveiling Ecological and Genetic Novelty within Lytic and Lysogenic Viral Communities of Hot Spring Phototrophic Microbial Mats. Microbiol. Spectr. 2021, 9, e00694-21. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 January 2017).
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Marker, A. The measurement of photosynthetic pigments in freshwaters and standardization of methods: Conclusions and recommendations. Arch. Hydrobiol. Beih. 1980, 14, 91–106. [Google Scholar]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Hong, C.; Manimaran, S.; Shen, Y.; Perez-Rogers, J.F.; Byrd, A.L.; Castro-Nallar, E.; Crandall, K.A.; Johnson, W.E. PathoScope 2.0: A complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2014, 2, 33. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Tully, B.J.; Graham, E.D.; Heidelberg, J.F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data 2018, 5, 170203. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Roux, S.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Huntemann, M.; Reddy, T.B.K.; Pons, J.C.; Llabrés, M. IMG/VR v. 2.0: An integrated data management and analysis system for cultivated and environmental viral genomes. Nucleic Acids Res. 2019, 47, D678–D686. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.G.Z.; Green, K.T.; Dutilh, B.E.; Edwards, R.A. SUPER-FOCUS: A tool for agile functional analysis of shotgun metagenomic data. Bioinformatics 2016, 32, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Yin, X.; Jiang, X.-T.; Chai, B.; Li, L.; Yang, Y.; Cole, J.R.; Tiedje, J.M.; Zhang, T. ARGs-OAP v2. 0 with an expanded SARG database and Hidden Markov Models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics 2018, 34, 2263–2270. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef]
- Liu, B.; Pop, M. ARDB—Antibiotic resistance genes database. Nucleic Acids Res. 2009, 37, D443–D447. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2013. [Google Scholar]
- Andersen, K.S.; Kirkegaard, R.H.; Karst, S.M.; Albertsen, M. ampvis2: An R package to analyse and visualise 16S rRNA amplicon data. bioRxiv 2018. [Google Scholar] [CrossRef]
- Tang, Y.; Horikoshi, M.; Li, W. ggfortify: Unified interface to visualize statistical results of popular R packages. R J. 2016, 8, 474. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Kassambara, A.; Kassambara, M.A. Package ‘ggpubr’; R Package Version 0.1; R Core Team: Vienna, Austria, 2020; Volume 6. [Google Scholar]
- Slowikowski, K.; Schep, A.; Hughes, S.; Lukauskas, S.; Irisson, J.-O.; Kamvar, Z.N.; Ryan, T.; Christophe, D.; Hiroaki, Y.; Gramme, P. Package ggrepel. Automatically Position Non-overlapping Text Labels with ‘ggplot2; R Core Team: Vienna, Austria, 2018. [Google Scholar]
- Auguie, B.; Antonov, A. gridExtra: Miscellaneous Functions for “grid” Graphics; R Core Team: Vienna, Austria, 2017; Volume 2. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Neuwirth, E.; Neuwirth, M.E. Package ‘RColorBrewer’. Phys. Rev. D—Part. Fields Gravit. Cosmol. 2011, 84, 1–5. [Google Scholar]
- Chamberlain, S.A.; Szöcs, E. taxize: Taxonomic search and retrieval in R. F1000Research 2013, 2, 191. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.A.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Ignacio-Espinoza, J.C.; Roux, S.; Doulcier, G.; Acinas, S.G.; Alberti, A.; Chaffron, S.; Cruaud, C.; De Vargas, C.; Gasol, J.M. Patterns and ecological drivers of ocean viral communities. Science 2015, 348, 1261498. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; Sullivan, M.B. The Pacific Ocean Virome (POV): A marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS ONE 2013, 8, e57355. [Google Scholar] [CrossRef] [PubMed]
- Nelson Silva, S. Progress in the Oceanographic Knowledge of Chilean Interior Waters, from Puerto Montt to Cape Horn; Comité Oceanográfico Nacional: Valparaíso, Chile, 2008. [Google Scholar]
- Chen, C.; Pan, J.; Xiao, S.; Wang, J.; Gong, X.; Yin, G.; Hou, L.; Liu, M.; Zheng, Y. Microplastics alter nitrous oxide production and pathways through affecting microbiome in estuarine sediments. Water Res. 2022, 221, 118733. [Google Scholar] [CrossRef] [PubMed]
- Jasmin, C.; Anas, A.; Singh, D.; Purohit, H.J.; Gireeshkumar, T.R.; Nair, S. Aberrations in the microbiome of cyanobacteria from a tropical estuary polluted by heavy metals. Mar. Pollut. Bull. 2020, 160, 111575. [Google Scholar] [CrossRef]
- Orel, N.; Fadeev, E.; Klun, K.; Ličer, M.; Tinta, T.; Turk, V. Bacterial indicators are ubiquitous members of pelagic microbiome in anthropogenically impacted coastal ecosystem. Front. Microbiol. 2022, 12, 765091. [Google Scholar] [CrossRef]
- Olsen, L.M.; Hernández, K.L.; Ardelan, M.V.; Iriarte, J.L.; Bizsel, K.C.; Olsen, Y. Responses in bacterial community structure to waste nutrients from aquaculture: An in situ microcosm experiment in a Chilean fjord. Aquac. Environ. Interact. 2017, 9, 21–32. [Google Scholar] [CrossRef]
- Dowle, E.; Pochon, X.; Keeley, N.; Wood, S.A. Assessing the effects of salmon farming seabed enrichment using bacterial community diversity and high-throughput sequencing. FEMS Microbiol. Ecol. 2015, 91, fiv089. [Google Scholar] [CrossRef]
- Duarte, L.N.; Coelho, F.J.; Cleary, D.F.; Bonifacio, D.; Martins, P.; Gomes, N.C. Bacterial and microeukaryotic plankton communities in a semi-intensive aquaculture system of sea bass (Dicentrarchus labrax): A seasonal survey. Aquaculture 2019, 503, 59–69. [Google Scholar] [CrossRef]
- Sun, M.; Zhan, Y.; Marsan, D.; Páez-Espino, D.; Cai, L.; Chen, F. Uncultivated Viral Populations Dominate Estuarine Viromes on the Spatiotemporal Scale. mSystems 2021, 6, e01020-20. [Google Scholar] [CrossRef]
- Alarcón-Schumacher, T.; Guajardo-Leiva, S.; Martinez-Garcia, M.; Díez, B. Ecogenomics and Adaptation Strategies of Southern Ocean Viral Communities. mSystems 2021, 6, e00396-21. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.Q.A.; Cabello, F.C.; L’Abée-Lund, T.M.; Tomova, A.; Godfrey, H.P.; Buschmann, A.H.; Sørum, H. Antimicrobial resistance and antimicrobial resistance genes in marine bacteria from salmon aquaculture and non-aquaculture sites. Environ. Microbiol. 2014, 16, 1310–1320. [Google Scholar] [CrossRef] [PubMed]
- Avendaño-Herrera, R.; Mancilla, M.; Miranda, C.D. Use of antimicrobials in Chilean Salmon farming: Facts, myths and perspectives. Rev. Aquac. 2023, 15, 89–111. [Google Scholar] [CrossRef]
- Salgado-Caxito, M.; Zimin-Veselkoff, N.; Adell, A.D.; Olivares-Pacheco, J.; Mardones, F.O. Qualitative Risk Assessment for Antimicrobial Resistance among Humans from Salmon Fillet Consumption Due to the High Use of Antibiotics against Bacterial Infections in Farmed Salmon. Antibiotics 2022, 11, 662. [Google Scholar] [CrossRef]
- Shade, A.; Peter, H.; Allison, S.; Baho, D.; Berga, M.; Buergmann, H.; Huber, D.; Langenheder, S.; Lennon, J.; Martiny, J.; et al. Fundamentals of Microbial Community Resistance and Resilience. Front. Microbiol. 2012, 3, 417. [Google Scholar] [CrossRef]
- Cuadrat, R.R.C.; Sorokina, M.; Andrade, B.G.; Goris, T.; Dávila, A.M.R. Global ocean resistome revealed: Exploring antibiotic resistance gene abundance and distribution in TARA Oceans samples. GigaScience 2020, 9, giaa046. [Google Scholar] [CrossRef]
- Fonseca, E.L.; Andrade, B.G.N.; Vicente, A.C.P. The Resistome of Low-Impacted Marine Environments Is Composed by Distant Metallo-β-Lactamases Homologs. Front. Microbiol. 2018, 9, 677. [Google Scholar] [CrossRef]
- Sutton, T.D.; Clooney, A.G.; Ryan, F.J.; Ross, R.P.; Hill, C. Choice of assembly software has a critical impact on virome characterisation. Microbiome 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zayed, A.A.; Conceição-Neto, N.; Temperton, B.; Bolduc, B.; Alberti, A.; Ardyna, M.; Arkhipova, K.; Carmichael, M.; Cruaud, C. Marine DNA viral macro-and microdiversity from pole to pole. Cell 2019, 177, 1109–1123.e14. [Google Scholar] [CrossRef]
- Tominaga, K.; Ogawa-Haruki, N.; Nishimura, Y.; Watai, H.; Yamamoto, K.; Ogata, H.; Yoshida, T. Prevalence of Viral Frequency-Dependent Infection in Coastal Marine Prokaryotes Revealed Using Monthly Time Series Virome Analysis. mSystems 2023, 8, e00931-22. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guajardo-Leiva, S.; Mendez, K.N.; Meneses, C.; Díez, B.; Castro-Nallar, E. A First Insight into the Microbial and Viral Communities of Comau Fjord—A Unique Human-Impacted Ecosystem in Patagonia (42∘ S). Microorganisms 2023, 11, 904. https://doi.org/10.3390/microorganisms11040904
Guajardo-Leiva S, Mendez KN, Meneses C, Díez B, Castro-Nallar E. A First Insight into the Microbial and Viral Communities of Comau Fjord—A Unique Human-Impacted Ecosystem in Patagonia (42∘ S). Microorganisms. 2023; 11(4):904. https://doi.org/10.3390/microorganisms11040904
Chicago/Turabian StyleGuajardo-Leiva, Sergio, Katterinne N. Mendez, Claudio Meneses, Beatriz Díez, and Eduardo Castro-Nallar. 2023. "A First Insight into the Microbial and Viral Communities of Comau Fjord—A Unique Human-Impacted Ecosystem in Patagonia (42∘ S)" Microorganisms 11, no. 4: 904. https://doi.org/10.3390/microorganisms11040904
APA StyleGuajardo-Leiva, S., Mendez, K. N., Meneses, C., Díez, B., & Castro-Nallar, E. (2023). A First Insight into the Microbial and Viral Communities of Comau Fjord—A Unique Human-Impacted Ecosystem in Patagonia (42∘ S). Microorganisms, 11(4), 904. https://doi.org/10.3390/microorganisms11040904