
Chromosome Segregation and Cell Division Defects in Escherichia coli Recombination Mutants Exposed to Different DNA-Damaging Treatments



Abstract
1. Introduction
2. Material and Methods
2.1. Bacterial Strains, Growth Media and Conditions
2.2. Measurement of Bacterial Survival after Introduction of DSBs by I-SceI Endonuclease
2.3. Fluorescence Microscopy of Bacterial Cells after Induction of I-SceI Expression
2.4. Measurement of Bacterial Survival after UV- and γ-Irradiation
2.5. Fluorescence Microscopy of UV- and γ-Irradiated Cells
2.6. Statistical Analyses
3. Results
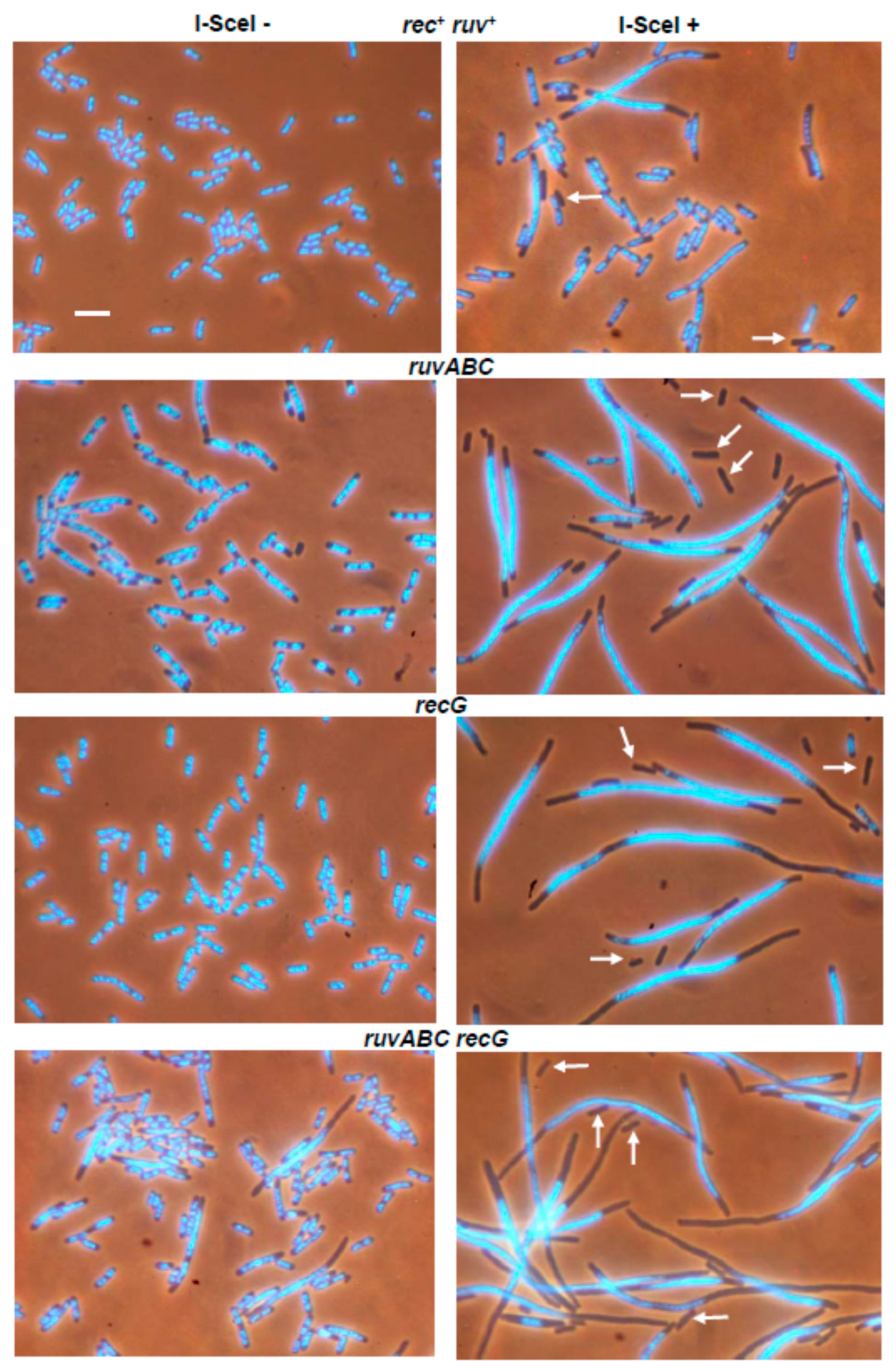
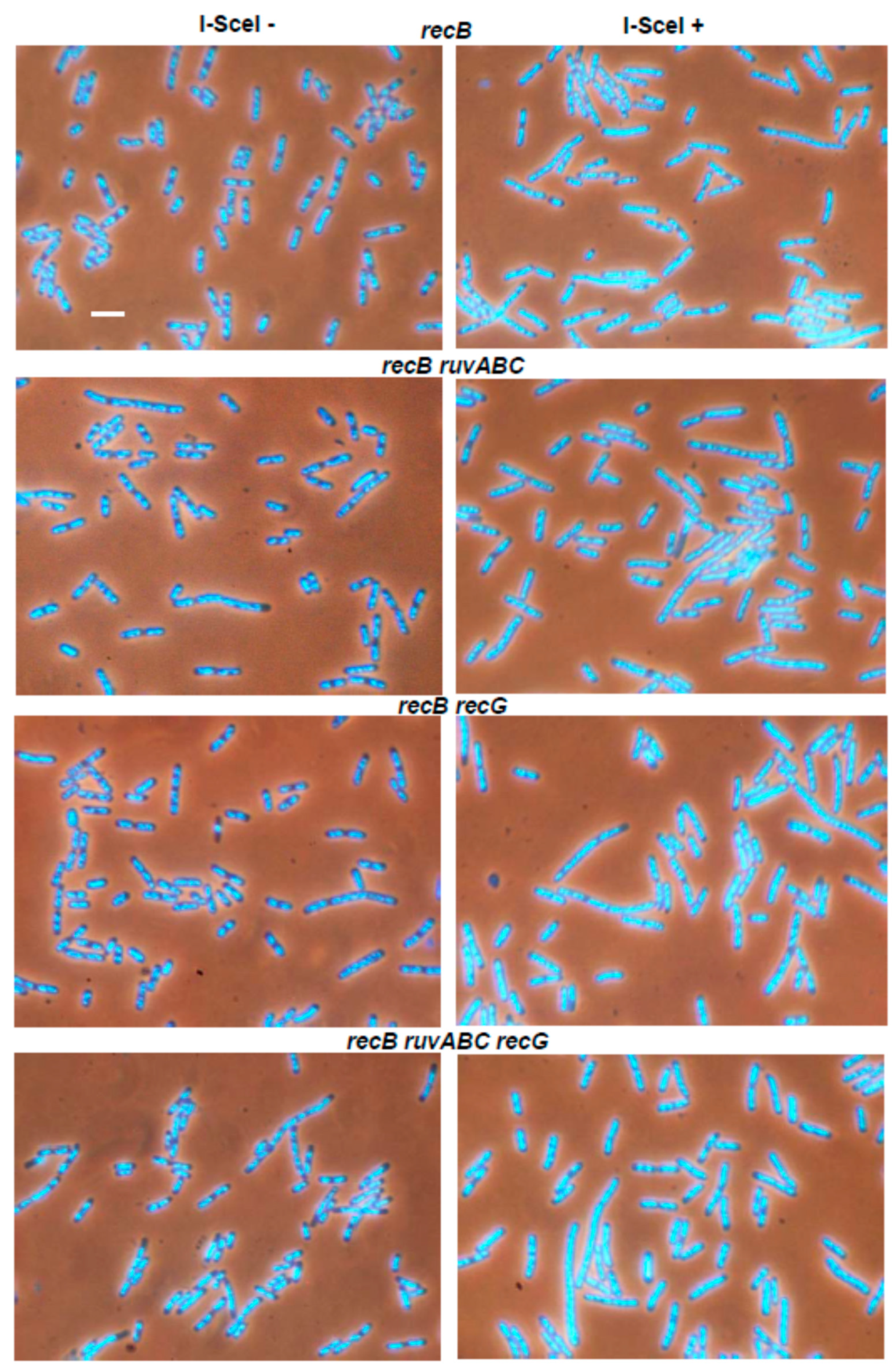
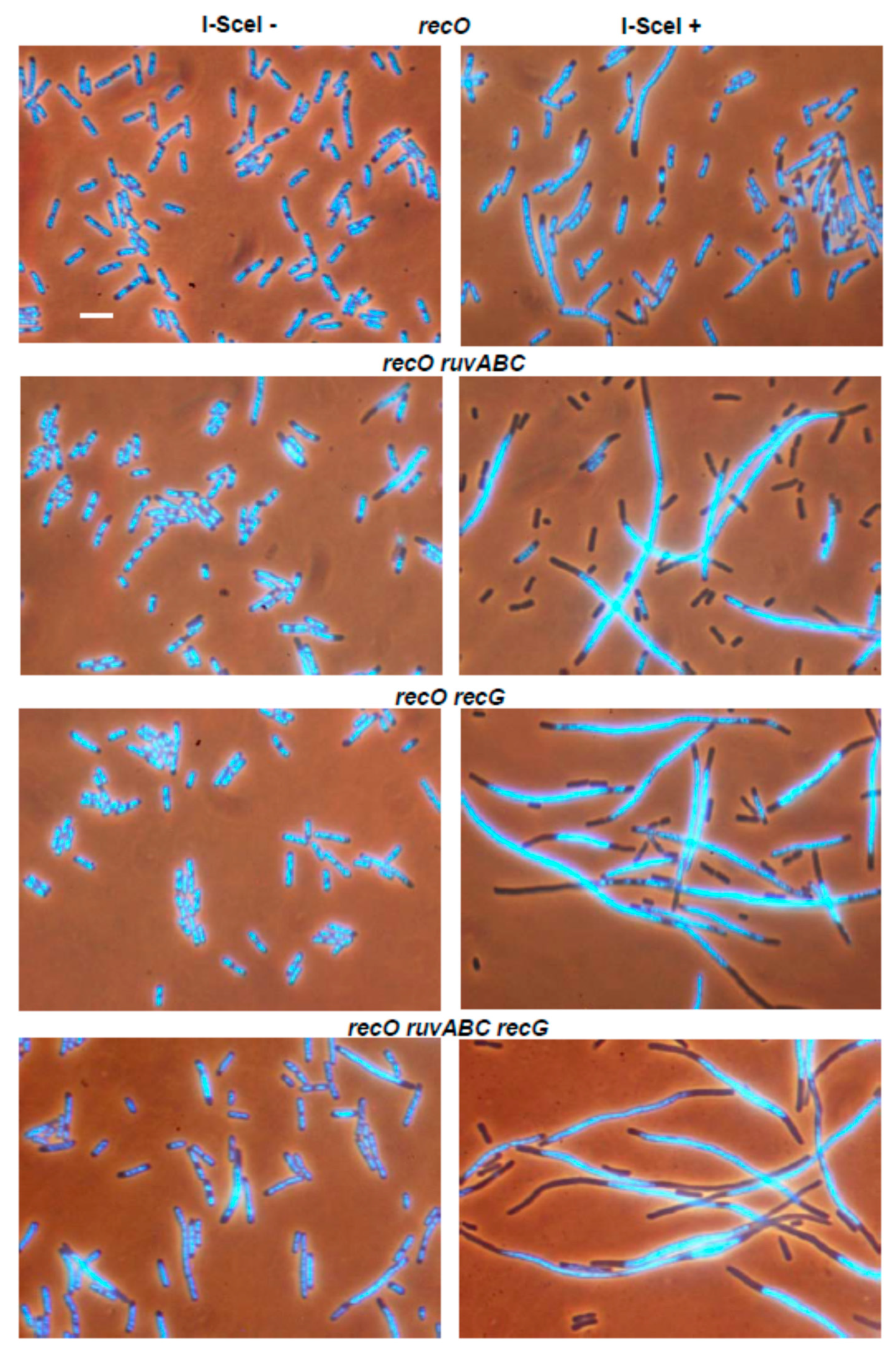
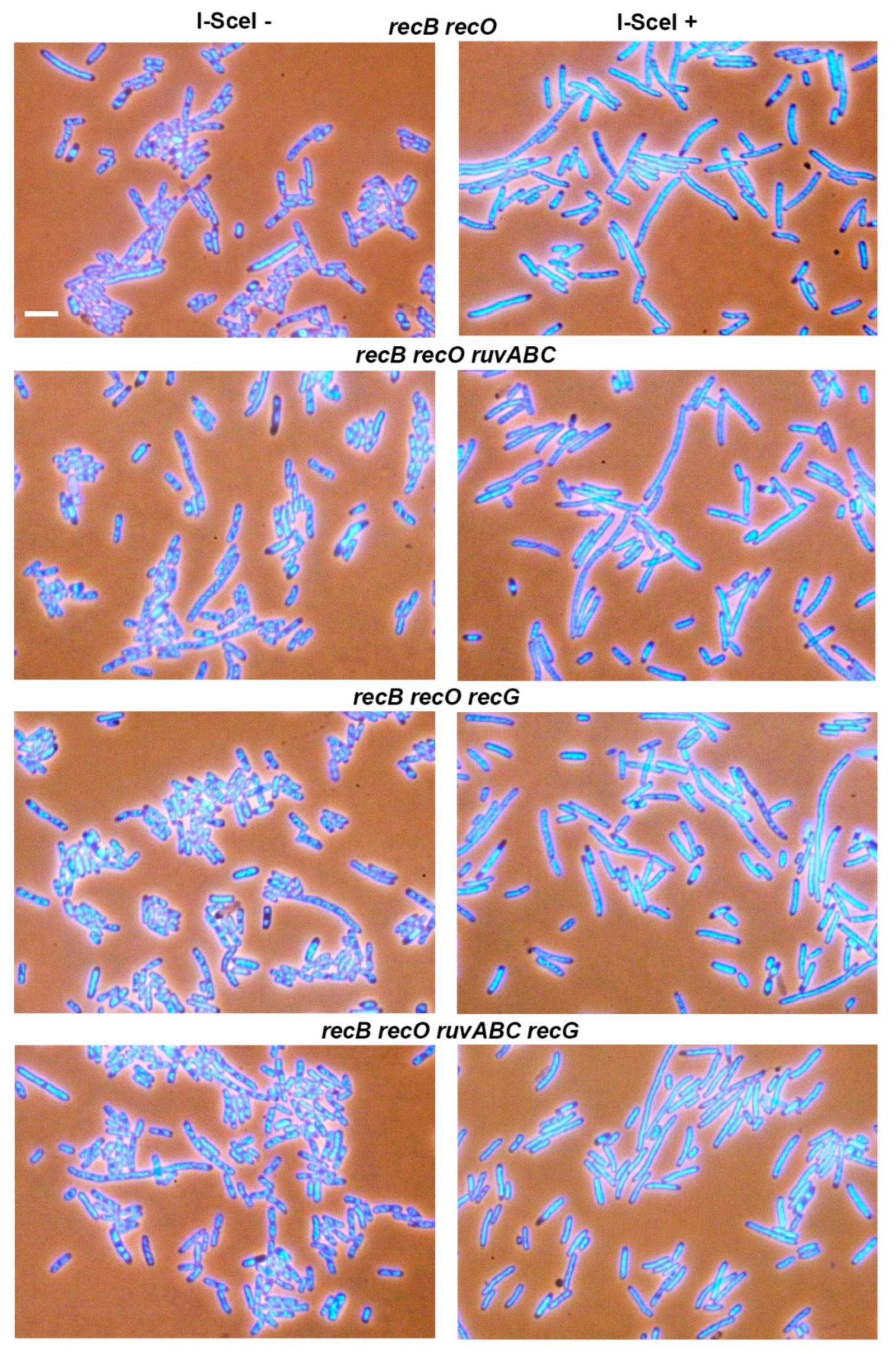
3.1. Effects of recB, recO, ruvABC and recG Mutations on Cell Survival and Morphology after Introduction of DSBs by I-SceI Endonuclease
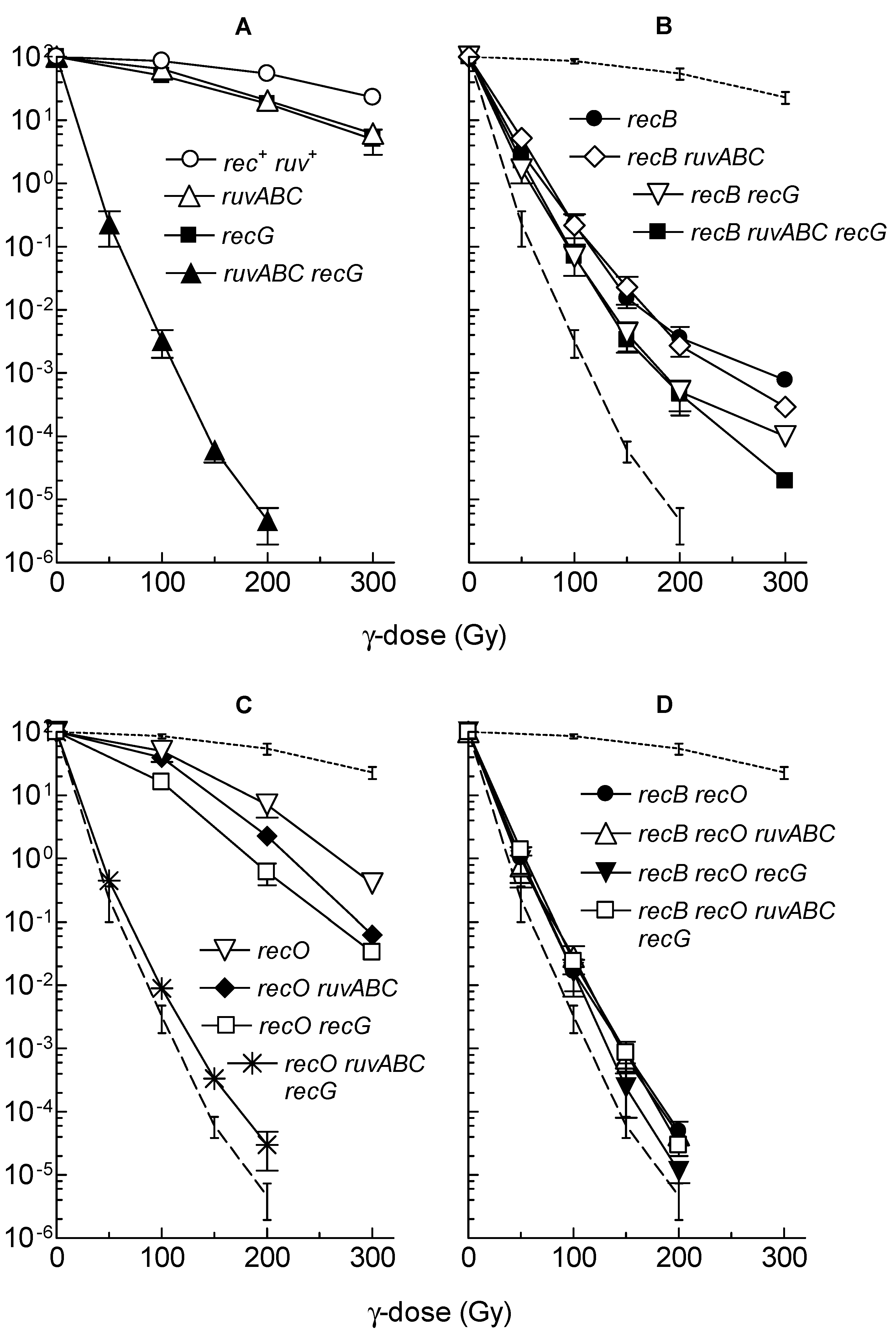
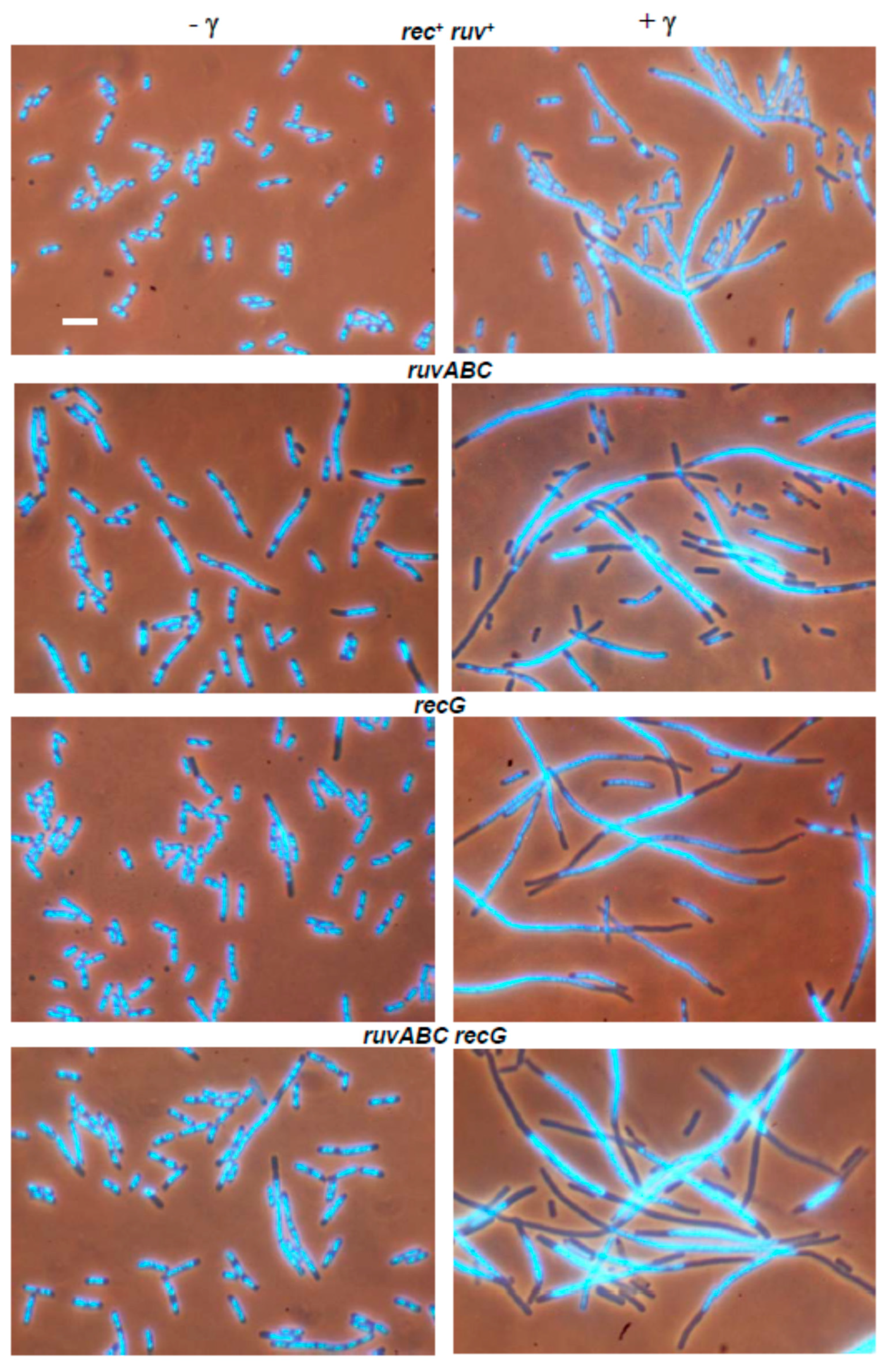
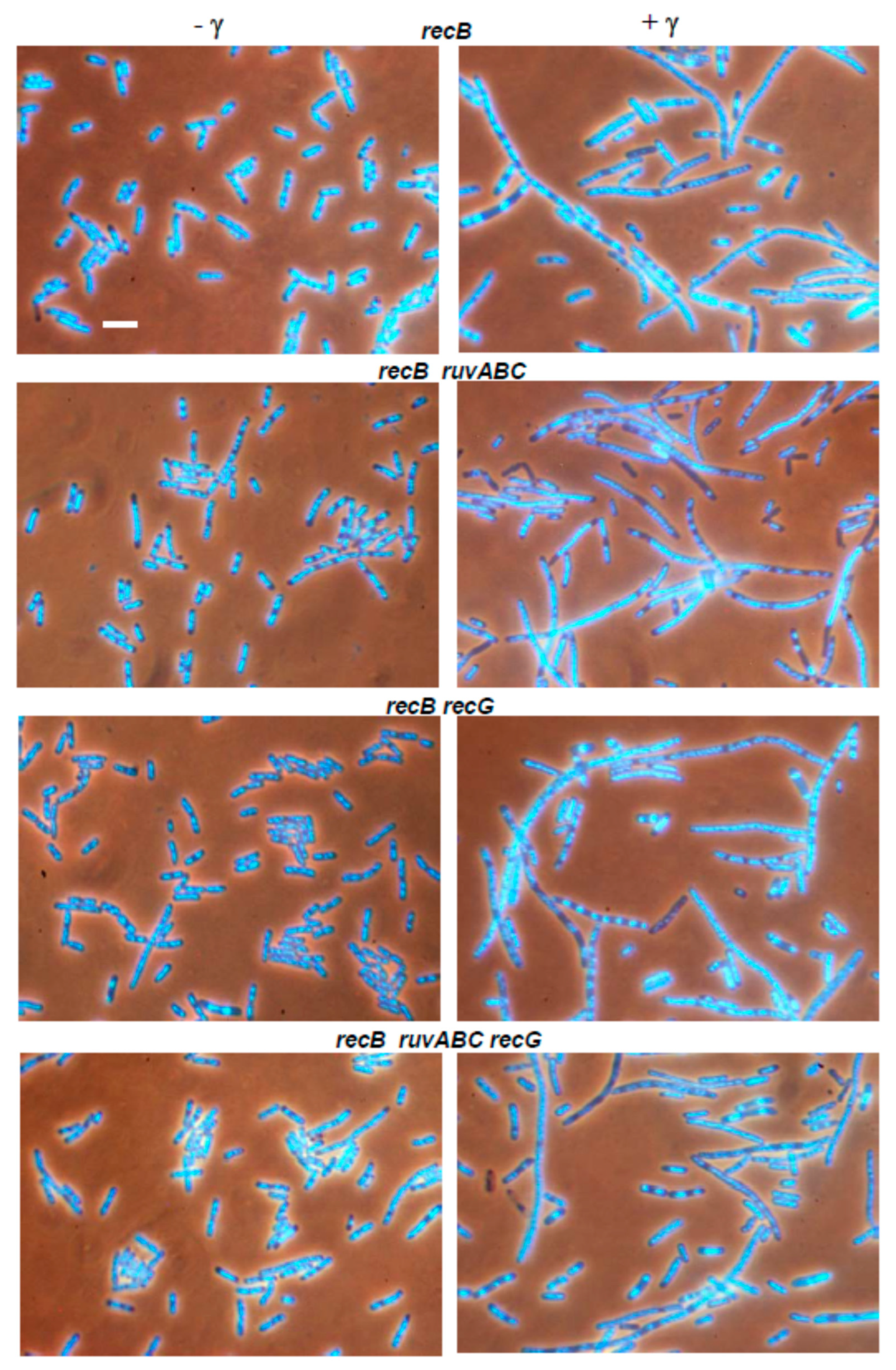
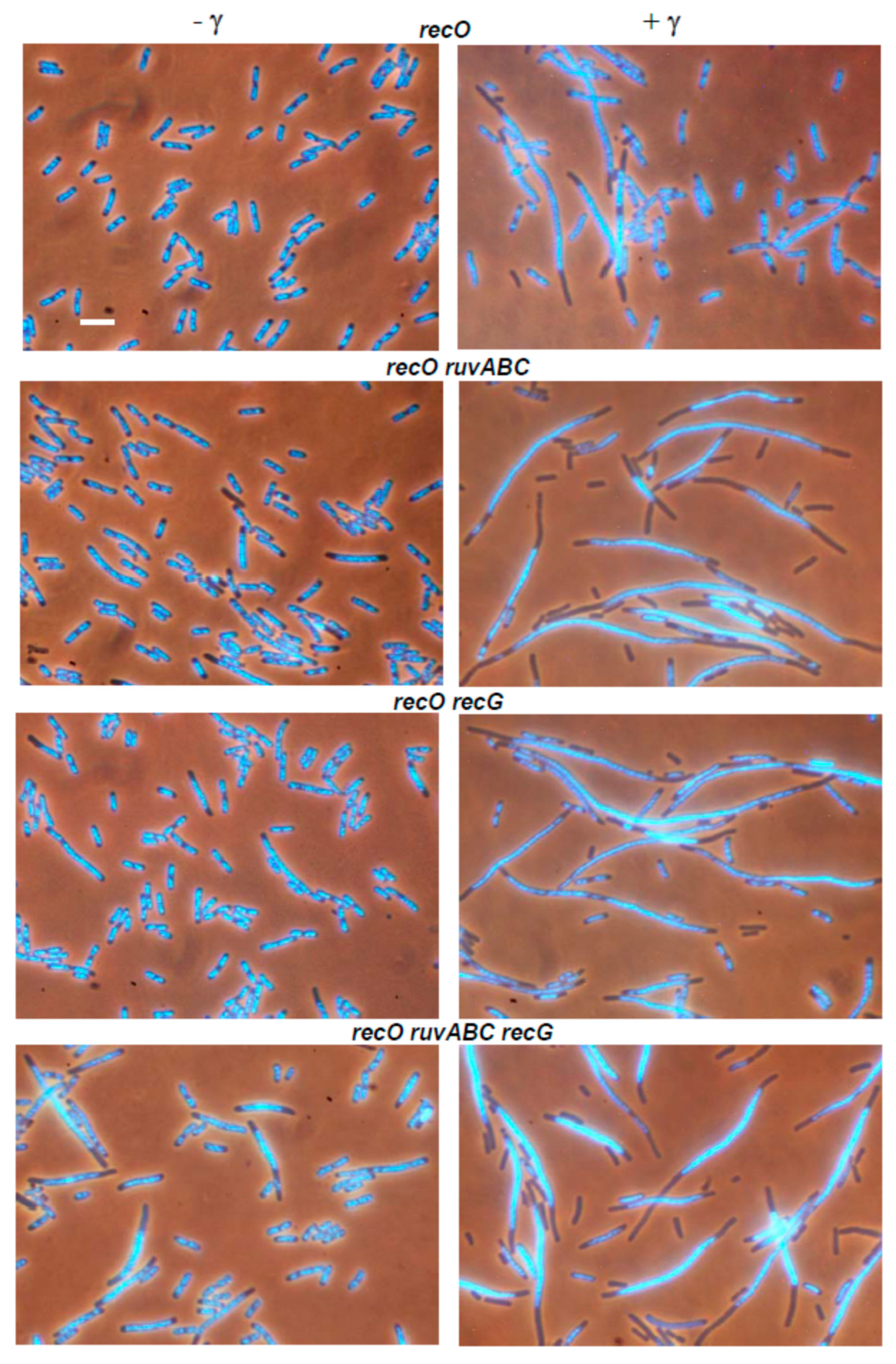
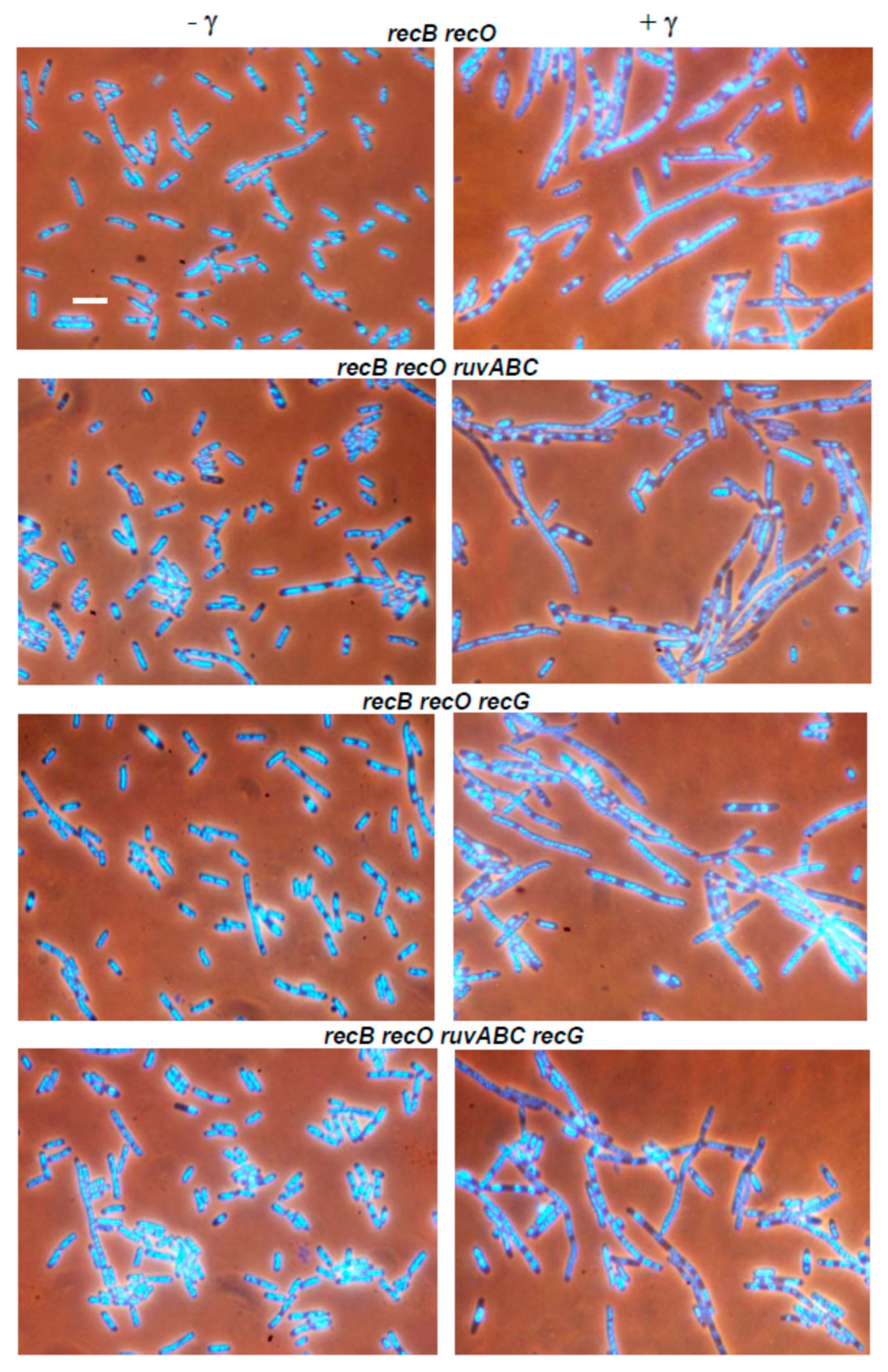
3.2. Effects of recB, recO, ruvABC and recG Mutations on Cell Survival and Morphology after Exposure to γ-Irradiation
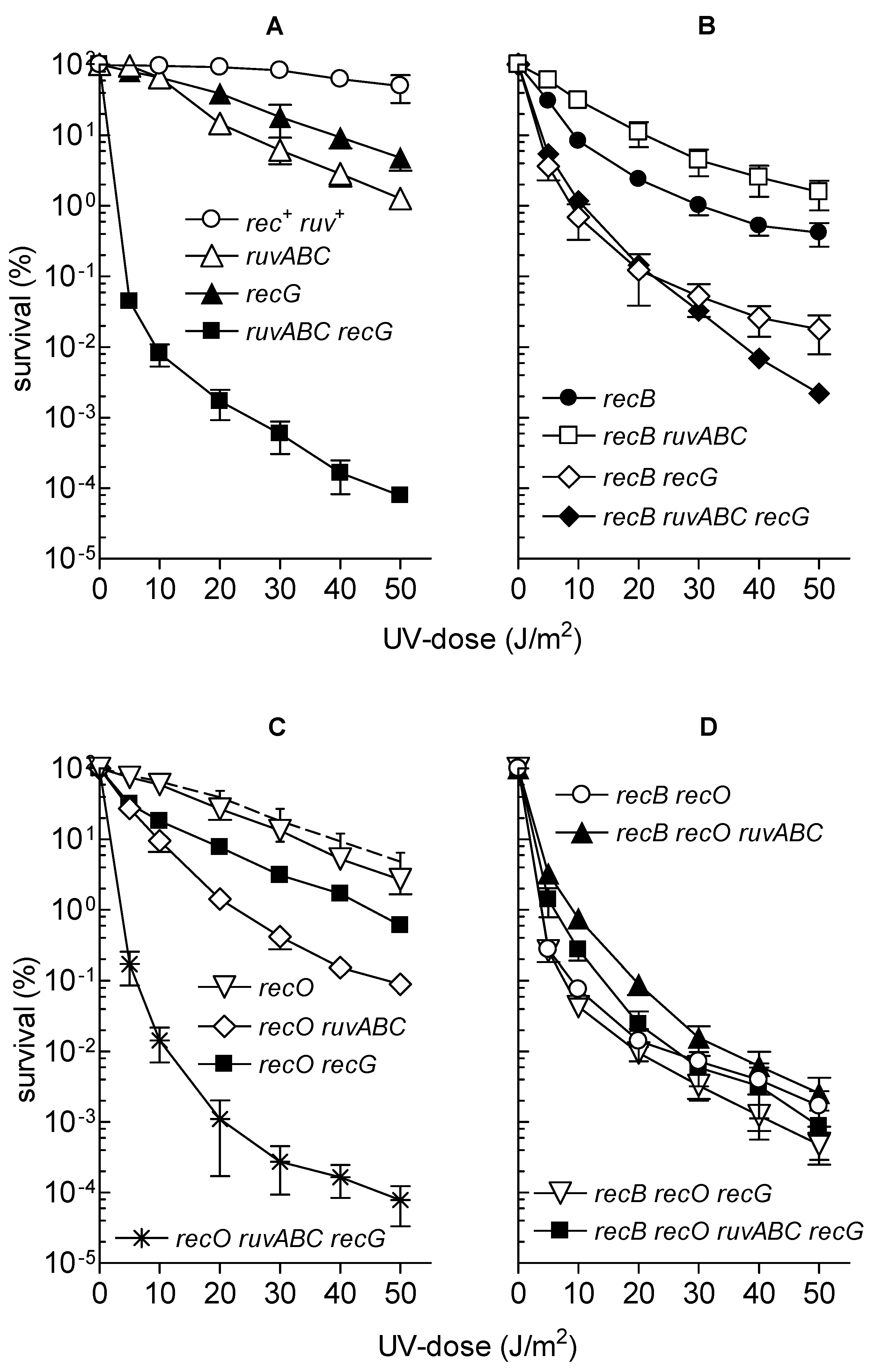
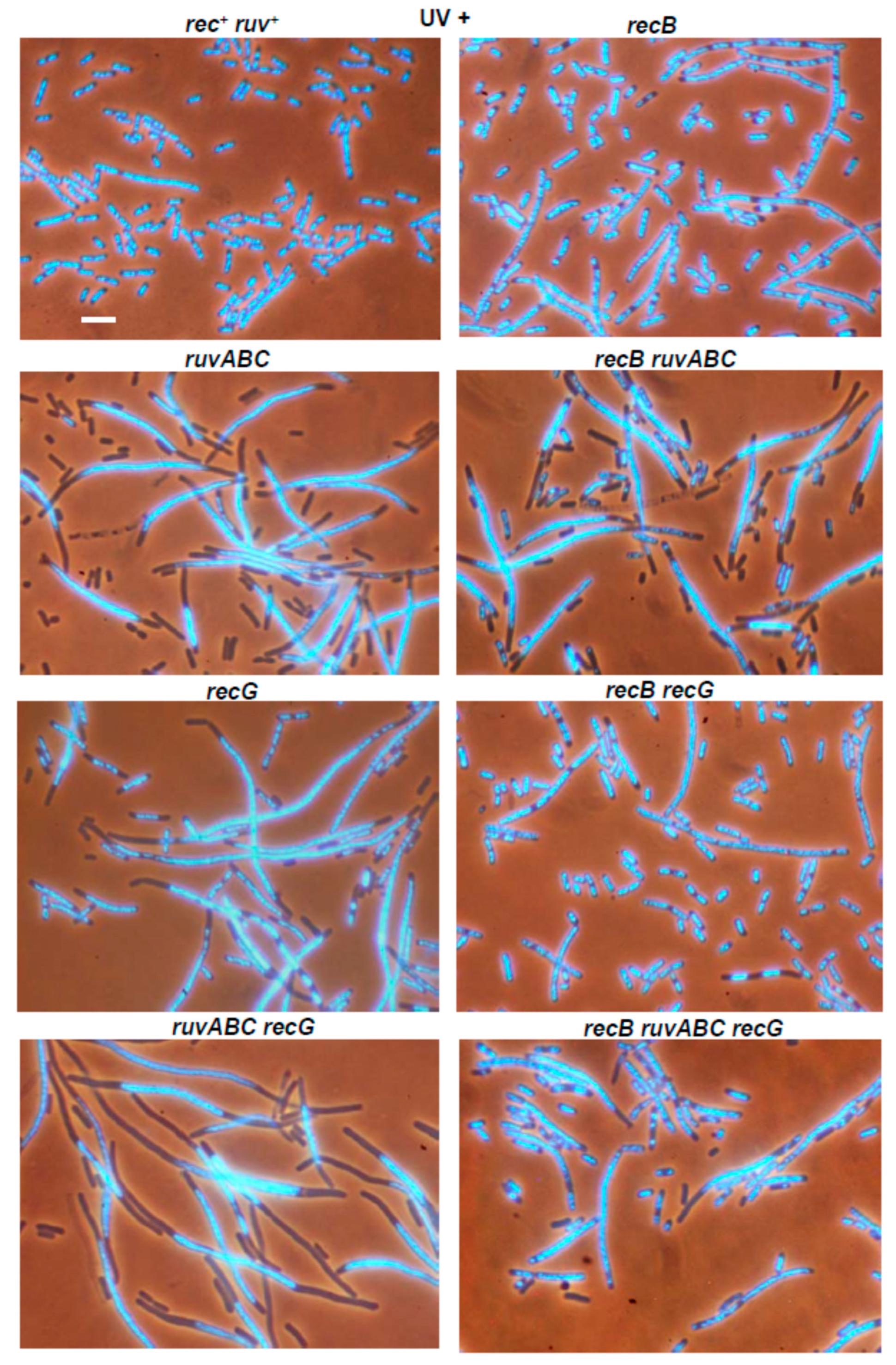
3.3. Effects of recB, recO, ruvABC and recG Mutations on Cell Survival and Morphology after Exposure to UV-Irradiation
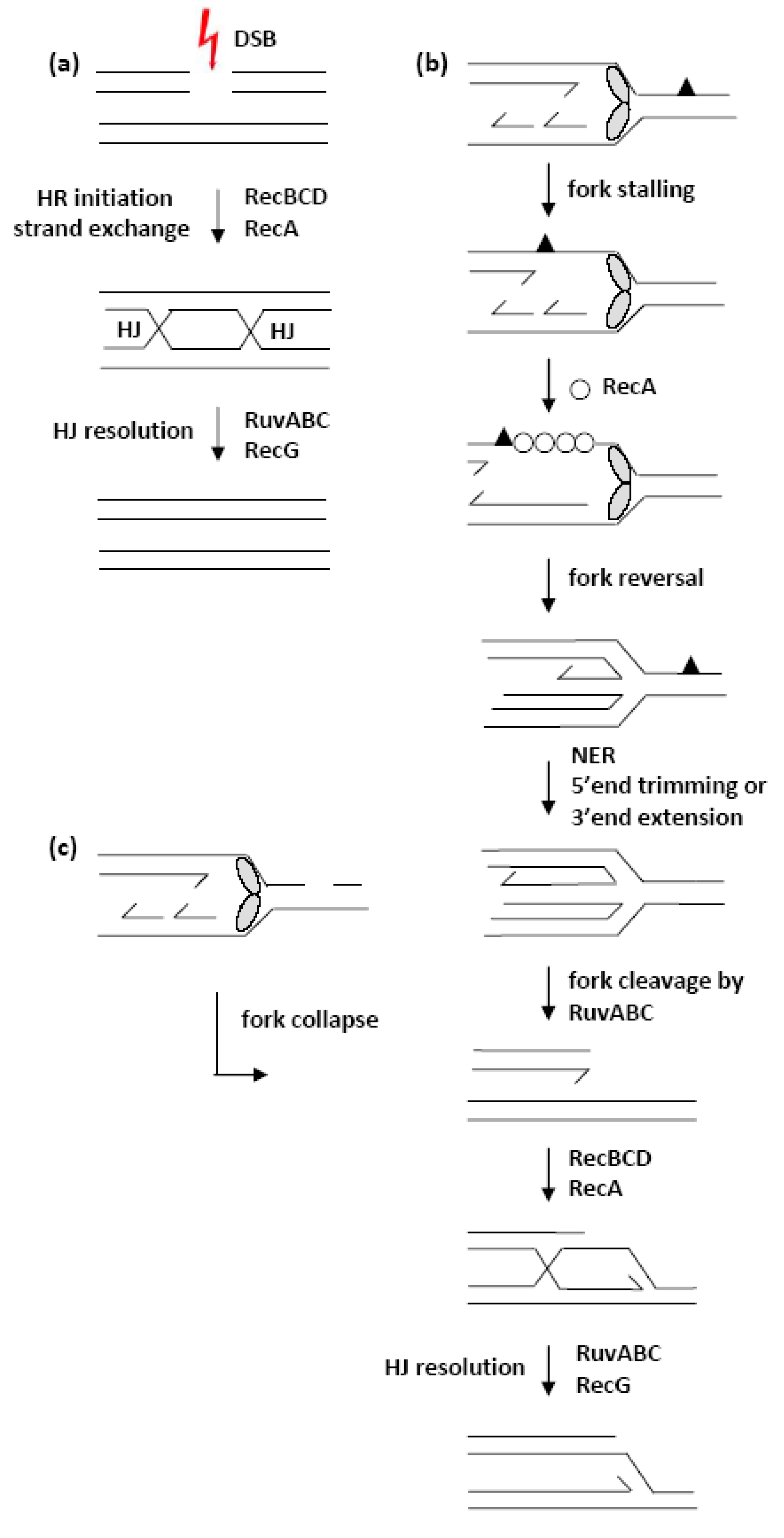
4. Discussion

Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spies, M.; Kowalczykowski, S.C. Homologous recombination by the RecBCD and RecF pathways. In The Bacterial Chromosome; Higgins, N.P., Ed.; ASM Press: Washington, DC, USA, 2005; pp. 389–403. [Google Scholar]
- Kuzminov, A. Homologous Recombination—Experimental Systems, Analysis and Significance. EcoSal Plus 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Leach, D. Homologous Recombination—Enzymes and Pathways. EcoSal Plus 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.M. Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 41–63. [Google Scholar] [CrossRef]
- West, S.C. Processing of recombination intermediates by the RuvABC proteins. Annu. Rev. Genet. 1997, 31, 213–244. [Google Scholar] [CrossRef]
- Lloyd, R.G. Conjugational recombination in resolvase-deficient ruvC mutants of Escherichia coli K-12 depends on recG. J. Bacteriol. 1991, 173, 5414–5418. [Google Scholar] [CrossRef]
- Meddows, T.R.; Savory, A.P.; Lloyd, R.G. RecG helicase promotes DNA double-strand break repair. Mol. Microbiol. 2004, 52, 119–132. [Google Scholar] [CrossRef]
- Rudolph, C.J.; Upton, A.L.; Briggs, G.S.; Lloyd, R.G. Is RecG a general guardian of the bacterial genome? DNA Repair. 2010, 9, 210–223. [Google Scholar] [CrossRef]
- Cox, M.M. Recombinational DNA repair of damaged replication forks in Escherichia coli: Questions. Annu. Rev. Genet. 2001, 35, 53–82. [Google Scholar] [CrossRef]
- Dillingham, M.S.; Kowalczykowski, S.C. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 2008, 72, 642–671. [Google Scholar] [CrossRef]
- Dixon, D.A.; Kowalczykowski, S.C. The recombination hotspot chi is a regulatory sequence that acts by attenuating the nuclease activity of the E. coli RecBCD enzyme. Cell 1993, 73, 87–96. [Google Scholar] [CrossRef]
- Anderson, D.G.; Kowalczykowski, S.C. The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in chi-regulated manner. Cell 1997, 90, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.A.; Kowalczykowski, S.C. Facilitated loading of RecA protein is essential to recombination by RecBCD enzyme. Genes Dev. 2000, 11, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Morimatsu, K.; Kowalczykowski, S.C. RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: A universal step of recombinational repair. Mol. Cell 2003, 11, 1337–1347. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Smith, K.C.; Wang, T.-C.V. recA-dependent DNA repair processes. Bioessays 1989, 10, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Kuzminov, A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol. Mol. Biol. Rev. 1999, 63, 751–813. [Google Scholar] [CrossRef]
- Persky, N.S.; Lovett, S.T. Mechanisms of recombination: Lessons from E. coli. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 347–370. [Google Scholar] [CrossRef]
- McGlynn, P.; Lloyd, R.G. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell 2000, 101, 35–45. [Google Scholar] [CrossRef]
- Khan, S.R.; Kuzminov, A. Replication forks stalled at ultraviolet lesions are rescued via RecA and RuvABC protein-catalyzed disintegration in Escherichia coli. J. Biol. Chem. 2012, 287, 6250–6265. [Google Scholar] [CrossRef]
- Bonura, T.; Smith, K.C. Enzymatic production of deoxyribonucleic acid double-strand breaks after ultraviolet irradiation of Escherichia coli K-12. J. Bacteriol. 1975, 121, 511–517. [Google Scholar] [CrossRef]
- Thoms, B.; Wackernagel, W. Interaction of RecBCD enzyme with DNA at double-strand breaks produced in UV-irradiated Escherichia coli: Requirement for DNA end processing. J. Bacteriol. 1998, 180, 5639–5645. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-C.V.; Smith, K.C. Mechanisms for recF-dependent and recB-dependent pathways of postreplication repair in UV-irradiated Escherichia coli uvrB. J. Bacteriol. 1983, 156, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-C.V.; Smith, K.C. Postreplicational formation and repair of DNA double-strand breaks in UV-irradiated Escherichia coli uvrB cells. Mutat. Res. 1986, 165, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.T.; Grossman, A.D.; Walker, G.C. Localization of UvrA and effect of DNA damage on the chromosome of Bacillus subtilis. J. Bacteriol. 2002, 184, 488–493. [Google Scholar] [CrossRef]
- Odsbu, I.; Skarstad, K. DNA compaction in the early part of the SOS response is dependent on RecN and RecA. Microbiology 2014, 160, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Ishioka, K.; Iwasaki, H.; Shinagawa, H. Roles of the recG gene product of Escherichia coli in recombination repair: Effects of the ΔrecG gene mutation on cell division and chromosome partition. Genes Genet. Syst. 1997, 72, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Ishioka, K.; Fukuoh, A.; Iwasaki, H.; Nakata, A.; Shinagawa, H. Abortive recombination in Escherichia coli ruv mutants blocks chromosome partitioning. Genes Cells 1998, 3, 209–220. [Google Scholar] [CrossRef]
- Buljubašić, M.; Zahradka, D.; Zahradka, K. RecQ helicase acts before RuvABC, RecG and XerC proteins during recombination in recBCD sbcBC mutants of Escherichia coli. Res. Microbiol. 2013, 164, 987–997. [Google Scholar] [CrossRef]
- Levin-Zaidman, S.; Frenkiel-Krispin, D.; Shimoni, E.; Sabanay, I.; Wolf, S.G.; Minsky, A. Ordered intracellular RecA-DNA assemblies: A potential site of in vivo RecA-mediated activities. Proc. Natl. Acad. Sci. USA 2000, 97, 6791–6796. [Google Scholar] [CrossRef]
- Zahradka, D.; Vlahović, K.; Petranović, M.; Petranović, D. Chromosome segregation and cell division defects in recBC sbcBC ruvC mutants of Escherichia coli. J. Bacteriol. 1999, 181, 6179–6183. [Google Scholar] [CrossRef]
- Xia, J.; Chen, L.-T.; Mei, Q.; Ma, C.-H.; Halliday, J.A.; Lin, H.Y.; Magnan, D.; Pribis, J.P.; Fitzgerald, D.M.; Hamilton, H.M.; et al. Holliday junction trap shows how cells use recombination and a junction-guardian role of RecQ helicase. Sci. Adv. 2016, 2, e1601605. [Google Scholar] [CrossRef]
- Bachmann, B.J. Derivations and genotypes of some mutant derivatives of Escherichia coli K-12. In Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd ed.; Neidhardt, F.C., Curtiss, R., Eds.; AMS Press: Washington, DC, USA, 1996; pp. 2460–2488. [Google Scholar]
- Miller, J.H. A Short Course in Bacterial Genetics; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1992. [Google Scholar]
- Vlašić, I.; Ivančić-Baće, I.; Imešek, M.; Mihaljević, B.; Brčić-Kostić, K. RecJ nuclease is required for SOS induction after introduction of a double-strand break in a RecA loading deficient recB mutant of Escherichia coli. Biochimie 2008, 90, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Vlašić, I.; Šimatović, A.; Brčić-Kostić, K. The hybrid recombinational repair pathway operates in a χ activity deficient recC1004 mutant of Escherichia coli. Biochimie 2012, 94, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Feliciello, I.; Zahradka, D.; Zahradka, K.; Ivanković, S.; Puc, N.; Đermić, D. RecF, UvrD, RecX and RecN proteins suppress DNA degradation at DNA double-strand breaks in Escherichia coli. Biochimie 2018, 148, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Buljubašić, M.; Hlevnjak, A.; Repar, J.; Đermić, D.; Filić, V.; Weber, I.; Zahradka, K.; Zahradka, D. RecBCD-RecFOR-independent pathway of homologous recombination in Escherichia coli. DNA Repair. 2019, 83, 102670. [Google Scholar] [CrossRef]
- Bolt, E.L.; Lloyd, R.G. Substrate specificity of RusA resolvase reveals the DNA structures targeted by RuvAB and RecG in vivo. Mol Cell 2002, 10, 187–198. [Google Scholar] [CrossRef]
- Gregg, A.V.; McGlynn, P.; Jaktaji, R.P.; Lloyd, R.G. Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol. Cell 2002, 9, 241–251. [Google Scholar] [CrossRef]
- Courcelle, J.; Hanawalt, P.C. RecA-dependent recovery of arrested DNA replication forks. Annu. Rev. Genet. 2003, 37, 611–646. [Google Scholar] [CrossRef]
- Rudolph, C.J.; Upton, A.L.; Lloyd, R.G. Maintaining replication fork integrity in UV-irradiated Escherichia coli cells. DNA Repair. 2008, 7, 1589–1602. [Google Scholar] [CrossRef]
- Michel, B.; Sinha, A.K.; Leach, D.R.F. Replication fork breakage and restart in Escherichia coli. Microbiol. Mol. Biol. Rev. 2018, 82, e00013-18. [Google Scholar] [CrossRef]
- Rothfield, L.; Taghbalout, A.; Shih, Y.-L. Spatial control of bacterial division-site placement. Nat. Rev. Microbiol. 2005, 3, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Raghunathan, S.; Chimthanawala, A.; Krishna, S.; Vecchiarelli, A.G.; Badrinarayanan, A. Asymmetric chromosome segregation and cell division in DNA damage-induced bacterial filaments. Mol. Biol. Cell 2020, 31, 2920–2931. [Google Scholar] [CrossRef] [PubMed]
- Vlašić, I.; Brčić-Kostić, K. Rescuing a sinking ship: The role of recombination gene products in SOS induction in Escherichia coli. Period. Biol. 2014, 116, 159–166. [Google Scholar]
- Ivančić-Baće, I.; Vlašić, I.; Salaj-Šmic, E.; Brčić-Kostić, K. Genetic evidence for the requirement of RecA loading activity in SOS induction after UV irradiation in Escherichia coli. J. Bacteriol. 2006, 188, 5024–5032. [Google Scholar] [CrossRef]
- Serment-Guerrero, J.; Breña-Valle, M.; Espinosa-Aguirre, J.J. In vivo role of Escherichia coli single-strand exonucleases in SOS induction by gamma radiation. Mutagenesis 2008, 23, 317–323. [Google Scholar] [CrossRef]
- Hegde, S.; Sandler, S.J.; Clark, A.J.; Madiraju, M.V.V.S. recO and recR mutants delay induction of the SOS response in Escherichia coli. Mol. Gen. Genet. 1995, 246, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Whitby, M.C.; Lloyd, R.G. Altered SOS induction associated with mutations in recF, recO and recR. Mol. Gen. Genet. 1995, 246, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.K.; Possoz, C.; Leach, D.R.F. The roles of bacterial double-strand break repair proteins in chromosomal DNA replication. FEMS Microbiol. Rev. 2020, 44, 351–368. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Relevant Genotype | Survival a (30 min) | Survival a (60 min) |
|---|---|---|---|
| TRM387 | rec+ ruv+ | 0.76 ± 0.099 | 0.39 ± 0.012 |
| LMM4199 | ΔruvABC | 0.68 ± 0.009 | 0.34 ± 0.101 |
| LMM4201 | ΔrecG | 0.19 ± 0.04 | 0.059 ± 0.006 |
| LMM4202 | ΔruvABC ΔrecG | 0.0054 ± 0.0029 | 0.00088 ± 0.00067 |
| LMM4198 | ΔrecB | 0.035 ± 0.011 | 0.0057 ± 0.0023 |
| LMM4611 | ΔrecB ΔruvABC | 0.039 ± 0.015 | 0.0061 ± 0.0017 |
| LMM4612 | ΔrecB ΔrecG | 0.036 ± 0.022 | 0.0071 ± 0.0035 |
| LMM4613 | ΔrecB ΔruvABC ΔrecG | 0.035 ± 0.0096 | 0.0042 ± 0.0011 |
| LMM4617 | ΔrecO | 0.72 ± 0.021 | 0.38 ± 0.045 |
| LMM4618 | ΔrecO ΔruvABC | 0.76 ± 0.05 | 0.49 ± 0.14 |
| LMM4619 | ΔrecO ΔrecG | 0.30 ± 0.06 | 0.14 ± 0.012 |
| LMM4620 | ΔrecO ΔruvABC ΔrecG | 0.0043 ± 0.0013 | 0.0011 ± 0.0002 |
| LMM4621 | ΔrecB ΔrecO | 0.013 ± 0.007 | 0.0037 ± 0.0016 |
| LMM4658 | ΔrecB ΔrecO ΔruvABC | 0.018 ± 0.009 | 0.0027 ± 0.0007 |
| LMM4659 | ΔrecB ΔrecO ΔrecG | 0.015 ± 0.0059 | 0.0023 ± 0.0014 |
| LMM4660 | ΔrecB ΔrecO ΔruvABC recG | 0.014 ± 0.0025 | 0.0047 ± 0.0018 |
| Strain | Relevant Genotype | Anucleate Cells (%) a,b | Filaments (%) a,c | ||
|---|---|---|---|---|---|
| I-SceI (-) d | I-SceI (+) d | I-SceI (-) d | I-SceI (+) d | ||
| TRM387 | rec+ ruv+ | 0 | 8.9 | 0.3 | 15.8 |
| LMM4199 | ΔruvABC | 1.0 | 54.5 | 9.3 | 29.5 |
| LMM4201 | ΔrecG | 0.1 | 28.8 | 1.7 | 33.1 |
| LMM4202 | ΔruvABC ΔrecG | 3.3 | 53.2 | 7.9 | 36.3 |
| LMM4198 | ΔrecB | 0 | 0 | 1.3 | 3.0 |
| LMM4611 | ΔrecB ΔruvABC | 0 | 0.3 | 6.1 | 8.1 |
| LMM4612 | ΔrecB ΔrecG | 0 | 0 | 2.2 | 3.7 |
| LMM4613 | ΔrecB ΔruvABC ΔrecG | 0 | 0 | 2.5 | 6.1 |
| LMM4617 | ΔrecO | 0 | 7.8 | 2.3 | 15.3 |
| LMM4618 | ΔrecO ΔruvABC | 1.2 | 50.3 | 8.8 | 23.8 |
| LMM4619 | ΔrecO ΔrecG | 0 | 24.3 | 3.4 | 24.7 |
| LMM4620 | ΔrecO ΔruvABC ΔrecG | 1.8 | 37.5 | 7.9 | 38.7 |
| LMM4621 | ΔrecB ΔrecO | 0.1 | 0.1 | 2.9 | 9.1 |
| LMM4658 | ΔrecB ΔrecO ΔruvABC | 0.1 | 0.3 | 5.0 | 10.4 |
| LMM4659 | ΔrecB ΔrecO ΔrecG | 0 | 0.1 | 3.3 | 11.8 |
| LMM4660 | ΔrecB ΔrecO ΔruvABC ΔrecG | 0.1 | 0.2 | 4.7 | 11.5 |
| Strain | Relevant Genotype | Annucleate Non-Irrad. | Cells (%) a,b γ-Irrad. d | UV-Irrad. e | Filaments Non-Irrad. | (%) a,c γ-Irrad. d | UV-Irrad. e |
|---|---|---|---|---|---|---|---|
| LMM2629 | rec+ ruv+ | 0 | 2.5 | 0.7 | 0.2 | 8.8 | 2.4 |
| LMM3188 | ruvABC | 0.7 | 58.3 | 76.4 | 4.6 | 18.6 | 18.1 |
| LMM3196 | recG | 0.1 | 3.7 | 20.2 | 1.6 | 31.9 | 25.6 |
| LMM3610 | ruvABC recG | 3.0 | 59.4 | 64.1 | 7.1 | 37.7 | 32.4 |
| LMM3183 | recB | 0 | 0 | 0 | 0.4 | 7.2 | 8.7 |
| LMM3625 | recB ruvABC | 0.2 | 11.3 | 43.5 | 3.4 | 19.1 | 17.7 |
| LMM3682 | recB recG | 0 | 0.3 | 0.1 | 0.8 | 6.1 | 10.5 |
| LMM4134 | recB ruvABC recG | 0 | 0.4 | 15.0 | 1.3 | 12.4 | 11.6 |
| LMM3545 | recO | 0.1 | 9.3 | 21.5 | 0.8 | 13.8 | 53.5 |
| LMM3600 | recO ruvABC | 1.2 | 61.4 | 35.3 | 2.5 | 21.2 | 52.0 |
| LMM3601 | recO recG | 0.2 | 14.7 | 21.9 | 3.5 | 25.0 | 60.7 |
| LMM3604 | recO ruvABC recG | 4.6 | 51.9 | 55.9 | 5.2 | 33.6 | 34.3 |
| LMM3599 | recB recO | 0.1 | 0.1 | 0.3 | 1.6 | 21.6 | 21.7 |
| LMM3602 | recB recO ruvABC | 0 | 0 | 1.2 | 2.1 | 20.3 | 21.0 |
| LMM3603 | recB recO recG | 0 | 0 | 0.4 | 2.2 | 21.2 | 21.8 |
| LMM3605 | recB recO ruvABC recG | 0.1 | 0.1 | 0.2 | 1.7 | 19.1 | 16.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahradka, K.; Repar, J.; Đermić, D.; Zahradka, D. Chromosome Segregation and Cell Division Defects in Escherichia coli Recombination Mutants Exposed to Different DNA-Damaging Treatments. Microorganisms 2023, 11, 701. https://doi.org/10.3390/microorganisms11030701
Zahradka K, Repar J, Đermić D, Zahradka D. Chromosome Segregation and Cell Division Defects in Escherichia coli Recombination Mutants Exposed to Different DNA-Damaging Treatments. Microorganisms. 2023; 11(3):701. https://doi.org/10.3390/microorganisms11030701
Chicago/Turabian StyleZahradka, Ksenija, Jelena Repar, Damir Đermić, and Davor Zahradka. 2023. "Chromosome Segregation and Cell Division Defects in Escherichia coli Recombination Mutants Exposed to Different DNA-Damaging Treatments" Microorganisms 11, no. 3: 701. https://doi.org/10.3390/microorganisms11030701
APA StyleZahradka, K., Repar, J., Đermić, D., & Zahradka, D. (2023). Chromosome Segregation and Cell Division Defects in Escherichia coli Recombination Mutants Exposed to Different DNA-Damaging Treatments. Microorganisms, 11(3), 701. https://doi.org/10.3390/microorganisms11030701