
Improved Quantitative Real-Time PCR Protocol for Detection and Quantification of Methanogenic Archaea in Stool Samples


Abstract
1. Introduction
2. Materials and Methods
2.1. Primer Design
2.2. DNA Templates
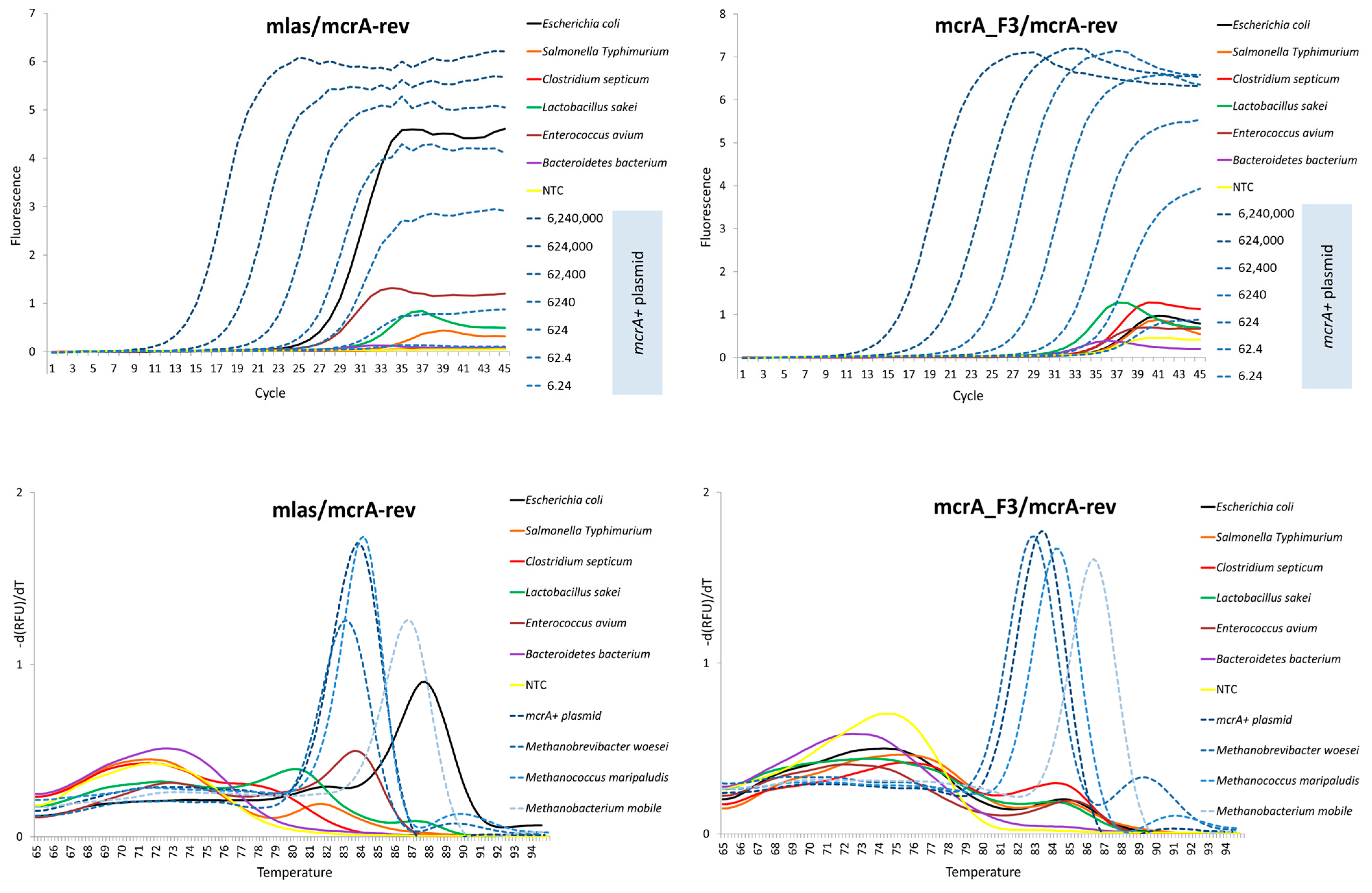
2.3. Initial Comparison of the Mlas and mcrA_F3 forward Primers
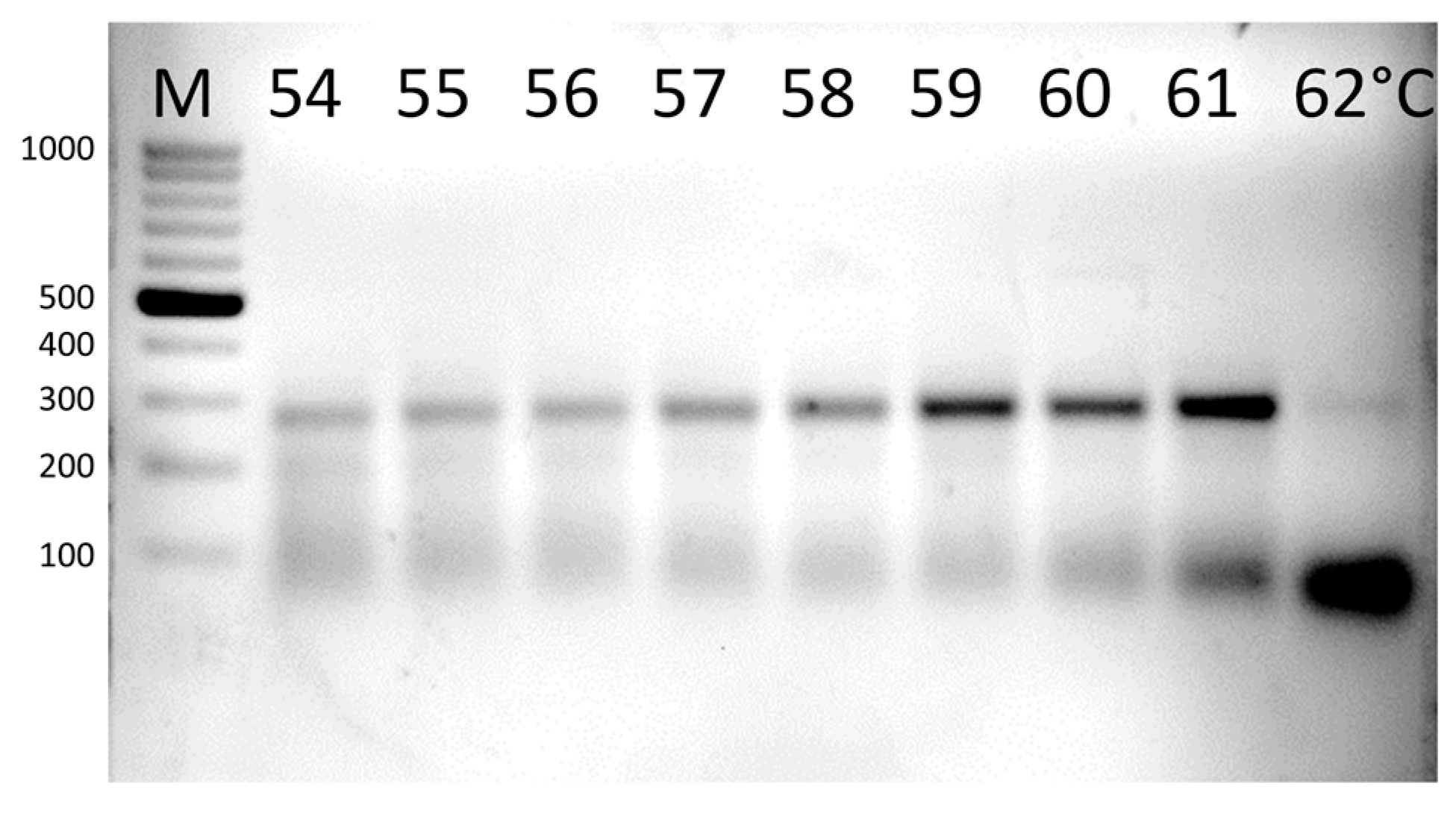
2.4. Gradient PCR
2.5. Selected Validation Parameters of the Assays
2.6. Detection of Methanogens in Chicken Dropping Samples
3. Results
3.1. Primer Design
3.2. Initial Comparison of the Mlas and mcrA_F3 Forward Primers
3.3. Gradient PCR
3.4. Selected Validation Parameters of the Assays
3.5. Methanogen Load in Chicken Dropping Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Djemai, K.; Drancourt, M.; Tidjani Alou, M. Bacteria and Methanogens in the Human Microbiome: A Review of Syntrophic Interactions. Microb. Ecol. 2022, 83, 536–554. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, X.; Zhang, T.; Si, H.; Xu, C.; Wright, A.D.G.; Li, G. Heterogeneous Development of Methanogens and the Correlation with Bacteria in the Rumen and Cecum of Sika Deer (Cervus Nippon) during Early Life Suggest Different Ecology Relevance. BMC Microbiol. 2019, 19, 129. [Google Scholar] [CrossRef]
- Sergeant, M.J.; Constantinidou, C.; Cogan, T.A.; Bedford, M.R.; Penn, C.W.; Pallen, M.J. Extensive Microbial and Functional Diversity within the Chicken Cecal Microbiome. PLoS ONE 2014, 9, e91941. [Google Scholar] [CrossRef] [PubMed]
- Brugère, J.F.; Borrel, G.; Gaci, N.; Tottey, W.; O’Toole, P.W.; Malpuech-Brugère, C. Archaebiotics: Proposed Therapeutic Use of Archaea to Prevent Trimethylaminuria and Cardiovascular Disease. Gut Microbes 2014, 5, 5–10. [Google Scholar] [CrossRef]
- Camara, A.; Konate, S.; Tidjani Alou, M.; Kodio, A.; Togo, A.H.; Cortaredona, S.; Henrissat, B.; Thera, M.A.; Doumbo, O.K.; Raoult, D.; et al. Clinical Evidence of the Role of Methanobrevibacter Smithii in Severe Acute Malnutrition. Sci. Rep. 2021, 11, 5426. [Google Scholar] [CrossRef]
- Lecours, P.B.; Marsolais, D.; Cormier, Y.; Berberi, M.; Haché, C.; Bourdages, R.; Duchaine, C. Increased Prevalence of Methanosphaera Stadtmanae in Inflammatory Bowel Diseases. PLoS ONE 2014, 9, e87734. [Google Scholar] [CrossRef] [PubMed]
- Moissl-Eichinger, C.; Pausan, M.; Taffner, J.; Berg, G.; Bang, C.; Schmitz, R.A. Archaea Are Interactive Components of Complex Microbiomes. Trends Microbiol. 2018, 26, 70–85. [Google Scholar] [CrossRef]
- Yeoman, C.J.; Chia, N.; Jeraldo, P.; Sipos, M.; Goldenfeld, N.D.; White, B.A. The Microbiome of the Chicken Gastrointestinal Tract. Anim. Health Res. Rev. 2012, 13, 89–99. [Google Scholar] [CrossRef]
- Liu, A.; Gao, W.; Zhu, Y.; Hou, X.; Chu, H. Gut Non-Bacterial Microbiota: Emerging Link to Irritable Bowel Syndrome. Toxins 2022, 14, 596. [Google Scholar] [CrossRef]
- Medvecky, M.; Cejkova, D.; Polansky, O.; Karasova, D.; Kubasova, T.; Cizek, A.; Rychlik, I. Whole Genome Sequencing and Function Prediction of 133 Gut Anaerobes Isolated from Chicken Caecum in Pure Cultures. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef]
- Segura-Wang, M.; Grabner, N.; Koestelbauer, A.; Klose, V.; Ghanbari, M. Genome-Resolved Metagenomics of the Chicken Gut Microbiome. Front. Microbiol. 2021, 12, 2390. [Google Scholar] [CrossRef] [PubMed]
- Saengkerdsub, S.; Anderson, R.C.; Wilkinson, H.H.; Kim, W.K.; Nisbet, D.J.; Ricke, S.C. Identification and Quantification of Methanogenic Archaea in Adult Chicken Ceca. Appl. Environ. Microbiol. 2007, 73, 353–356. [Google Scholar] [CrossRef]
- Saengkerdsub, S.; Herrera, P.; Woodward, C.L.; Anderson, R.C.; Nisbet, D.J.; Ricke, S.C. Detection of Methane and Quantification of Methanogenic Archaea in Faeces from Young Broiler Chickens Using Real-Time PCR. Lett. Appl. Microbiol. 2007, 45, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Cisek, A.A.; Bąk, I.; Stefańska, I.; Binek, M. Selection and Optimization of High-Yielding DNA Isolation Protocol for Quantitative Analyses of Methanogenic Archaea. Microorganisms 2022, 10, 523. [Google Scholar] [CrossRef] [PubMed]
- Narihiro, T.; Sekiguchi, Y. Oligonucleotide Primers, Probes and Molecular Methods for the Environmental Monitoring of Methanogenic Archaea. Microb. Biotechnol. 2011, 4, 585–602. [Google Scholar] [CrossRef]
- The Ribosomal RNA Database. Available online: https://rrndb.umms.med.umich.edu/ (accessed on 24 November 2021).
- WebPRANK. Available online: https://www.ebi.ac.uk/goldman-srv/webprank/ (accessed on 14 January 2022).
- Steinberg, L.M.; Regan, J.M. McrA-Targeted Real-Time Quantitative PCR Method to Examine Methanogen Communities. Appl. Environ. Microbiol. 2009, 75, 4435–4442. [Google Scholar] [CrossRef] [PubMed]
- Oligo Analyzer. Available online: https://www.idtdna.com/calc/analyzer (accessed on 12 January 2021).
- Primer-BLAST. Available online: https://www.ncbi.nlm.nih.gov/tools/primer-blast/ (accessed on 14 January 2021).
- Steinberg, L.M.; Regan, J.M. Phylogenetic Comparison of the Methanogenic Communities from an Acidic, Oligotrophic Fen and an Anaerobic Digester Treating Municipal Wastewater Sludge. Appl. Environ. Microbiol. 2008, 74, 6663–6671. [Google Scholar] [CrossRef]
- Copy Number Calculator for Realtime PCR|Science Primer. Available online: http://scienceprimer.com/copy-number-calculator-for-realtime-pcr (accessed on 24 May 2019).
- Wang, Q.; Wang, X.; Zhang, J.; Song, G. LNA Real-Time PCR Probe Quantification of Hepatitis B Virus DNA. Exp. Ther. Med. 2012, 3, 503–508. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, R.; Ou, X.; Yao, D.; Huang, Z.; Li, L.; Sun, B. Primers and Probe Design and Precision Assessment of the Real Time RT-PCR Assay in Coxsackievirus A10 and Enterovirus Detection. Data Br. 2017, 12, 418–422. [Google Scholar] [CrossRef]
- Lin, C.N.; Lin, W.H.; Hung, L.N.; Wang, S.Y.; Chiou, M.T. Comparison of Viremia of Type II Porcine Reproductive and Respiratory Syndrome Virus in Naturally Infected Pigs by Zip Nucleic Acid Probe-Based Real-Time PCR. BMC Vet. Res. 2013, 9, 814–822. [Google Scholar] [CrossRef]
- Chen, Y.; Zhen, Y.; Wan, J.; Yin, X.; Li, S.; Liu, J.; Zhang, G.; Mi, T. Differences in the Methanogen Community between the Nearshore and Offshore Sediments of the South Yellow Sea. J. Microbiol. 2022, 60, 814–822. [Google Scholar] [CrossRef]
- Buessecker, S.; Zamora, Z.; Sarno, A.F.; Finn, D.R.; Hoyt, A.M.; van Haren, J.; Urquiza Muñoz, J.D.; Cadillo-Quiroz, H. Microbial Communities and Interactions of Nitrogen Oxides With Methanogenesis in Diverse Peatlands of the Amazon Basin. Front. Microbiol. 2021, 12, 659079. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.G.; Logroño, W.; da Rocha, U.N.; Harms, H.; Nikolausz, M. Enrichment of Anaerobic Microbial Communities from Midgut and Hindgut of Sun Beetle Larvae (Pachnoda Marginata) on Wheat Straw: Effect of Inoculum Preparation. Microorganisms 2022, 10, 761. [Google Scholar] [CrossRef] [PubMed]
- Teigen, L.; Mathai, P.P.; Matson, M.; Lopez, S.; Kozysa, D.; Kabage, A.J.; Hamilton, M.J.; Vaughn, B.P.; Sadowsky, M.J.; Khoruts, A. Methanogen Abundance Thresholds Capable of Differentiating In Vitro Methane Production in Human Stool Samples. Dig. Dis. Sci. 2021, 66, 3822–3830. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligo Name | Sequence 5′–3′ | Oligo Binding Site 5′–3′ * | Tm [°C] | Product Size with Primer mcrA-Rev [bp] | References |
|---|---|---|---|---|---|
| mcrA-rev | CGTTCATBGCGTAGTTVGGRTAGT | 446–467 | 62–68 | n/a | [21] |
| mlas | GGTGGTGTMGGDTTCACMCARTA | 1–23 | 62–70 | 469 | [21] |
| mcrA_F3 | CTTGAARMTCACTTCGGTGGWTC | 199–221 | 62–66 | 271 | This study and [14] |
| Acc. No. | Species | mcrA_F3 Sequence CTTGAARMTCACTTCGGTGGWTC | Product Size with Primer mcrA-Rev (bp) * |
|---|---|---|---|
| KC618341.1 | Methanopyrus sp. | ATGGAGACCCACTTCGGTGGATC | 271 |
| HQ896500.1 | Methanomassiliicoccus luminyensis | ATGGAAACCCACTTCGGTGGTTC | 271 |
| AF414042.1 | Methanopyrus kandleri | ATGGAGACGCACTTCGGAGGTTC | 271 |
| MH004454.1 | Methanosarcina mazei | CTTGAAGACCACTTCGGTGGGTC | 271 |
| AB288266.1 | Methanosarcina horonobensis | CTTGAAGACCACTTCGGTGGATC | 271 |
| AB288268.1 | Methanosarcina subterranea | CTTGAAGACCACTTCGGTGGGTC | 271 |
| U22242.1 | Methanolobus oregonensis | CTTGAGGACCACTTCGGTGGATC | n/a |
| EU715818.1 | Methanolobus zinderi | CTCGAAGACCACTTCGGTGGATC | 271 |
| U22243.1 | Methanolobus taylorii | CTCGAAGACCACTTTGGTGGATC | n/a |
| AB703629.1 | Methanolobus profundi | CTCGAAGACCACTTCGGTGGATC | n/a |
| U22245.1 | Methanolobus vulcani | CTCGAAGACCACTTCGGTGGATC | n/a |
| JN081865.1 | Methanocella conradii | CTCGAGGACCACTTCGGCGGCTC | n/a |
| KJ441441.1 | Methanocella sp. | CTCGAGGACCACTTCGGTGGCTC | n/a |
| AB300467.1 | Methanocella paludicola | CTCGAGGACCACTTCGGTGGCTC | n/a |
| AF414044.1 | Methanomicrobium mobile | ATGGAAGACCACTTCGGCGGTTC | n/a |
| MH004450.1 | Methanoculleus marisnigri | ATGGAGGACCACTTCGGCGGTTC | 271 |
| AB300784.1 | Methanoculleus palmolei | ATGGAGGACCACTTCGGCGGGTC | n/a |
| AB300787.1 | Methanoculleus bourgensis | ATGGAAGACCACTTCGGCGGGTC | 271 |
| AB300779.1 | Methanoculleus chikugoensis | ATGGAGGACCACTTCGGCGGTTC | 271 |
| DQ229160.1 | Methanogenium organophilum | ATGGAAGACCACTTCGGTGGTTC | 271 |
| DQ229161.1 | Methanogenium boonei | ATGGAAGACCACTTTGGCGGGTC | n/a |
| JQ917190.1 | Methanobacterium sp. | CTGGAAGACCACTTTGGCGGTTC | n/a |
| GU385700.1 | Methanobrevibacter smithii | CTTGAAACTCACTTCGGTGGATC | 271 |
| LK054628.1 | Methanobrevibacter oralis | CTCGAAACTCACTTCGGTGGATC | n/a |
| CP001719.1 | Methanobrevibacter ruminantium | CTTGAAACTCACTTCGGTGGTTC | 271 |
| KF214818.1 | Methanobrevibacter sp. D5/mcrA+ plasmid | CTTGAAACTCACTTCGGTGGATC | 271 |
| EU919431.1 | Methanobrevibacter gottschalkii | CTTGAAACTCACTTCGGTGGATC | 271 |
| KC865050.1 | Methanobrevibacter boviskoreani | CTTGAAACCCAATTCGGTGGATC | 271 |
| EU919432.1 | Methanobrevibacter woesei | CTTGAAACTCAATTCGGTGGATC | 271 |
| KC865051.1 | Methanobrevibacter wolinii | CTTGAAACTCAATTCGGTGGATC | 271 |
| AF414035.1 | Methanobrevibacter arboriphilus | CTTGAAACCCAATTCGGTGGTTC | n/a |
| HM802934.1 | Methanobacterium movens | TTAGAAACCCTCTTCGGTGGATC | n/a |
| AY289750.1 | Methanobacterium thermaggregans | CTCGAGGACCAGTTTGGTGGATC | n/a |
| AB300780.1 | Methanothermobacter wolfeii | CTGGAGGACCAGTTCGGAGGATC | 271 |
| AB523786.1 | Methanothermobacter tenebrarum | CTCGAGACACAATTTGGAGGATC | 271 |
| HQ283274.1 | Methanothermobacter crinale | CTTGAGACACAGTTCGGCGGATC | n/a |
| KP006500.1 | Methanobacterium aggregans | CTCGAGACCCAGTTCGGTGGATC | n/a |
| AY386125.1 | Methanobacterium aarhusense | TTAGAAACACAGTTCGGTGGATC | n/a |
| AF313806.1 | Methanobacterium bryantii | CTTGAAGATCAGTTCGGTGGATC | n/a |
| X07793.1 | Methanococcus voltae | TTAGAAGACCACTTCGGTGGCTC | 271 |
| AF414048.1 | Methanothermococcus thermolithotrophicus | TTAGAAGACCACTTCGGAGGTTC | 271 |
| AB703637.1 | Methanococcus maripaludis | TTAGAAGACCACTTCGGTGGATC | 271 |
| M16893.1 | Methanococcus vannielii | TTAGAAGACCACTTTGGTGGATC | 271 |
| AY354034.1 | Methanococcus aeolicus | TTGGAAGACCACTTTGGAGGTTC | 271 |
| FJ982887.1 | Methanosphaera sp. | TTAGAAGACCACTTCGGTGGATC | 271 |
| AF414047.1 | Methanosphaera stadtmanae | TTAGAAGACCACTTTGGTGGATC | 271 |
| Estimated Plasmid/Genome Copy No. | mlas/mcrA-Rev | mcrA_F3/mcrA-Rev | ||||
|---|---|---|---|---|---|---|
| No. Positive/No. Tested | Melting Analysis * | No. Positive/No. Tested | Melting Analysis * | |||
| mcrA+ Plasmid | ||||||
| 6.24 × 106 | 8/8 | S | 9/9 | S | ||
| 6.24 × 105 | 8/8 | 9/9 | ||||
| 6.24 × 104 | 8/8 | 9/9 | ||||
| 6.24 × 103 | 8/8 | 9/9 | ||||
| 6.24 × 102 | 8/8 | 10/10 | ||||
| 6.24 × 101 | 8/8 | 6/8 | 10/10 | |||
| 2/8 | S + PD | |||||
| 6.24 × 100 | 2/8 | S + PD | 3/10 | S + PD | ||
| 6.24 × 10−1 | 0/8 | PD | 0/10 | PD | ||
| Methanobrevibacter woesei DSM 11979 | ||||||
| 6 × 106 | n/a | n/a | 2/2 | S | ||
| 6 × 105 | 8/8 | S | 7/7 | |||
| 6 × 104 | 8/8 | 7/7 | ||||
| 6 × 103 | 8/8 | 7/7 | ||||
| 6 × 102 | 8/8 | 8/8 | ||||
| 6 × 101 | 8/8 | 2/8 | 8/8 | 4/8 | ||
| 6/8 | S + PD | 4/8 | S + PD | |||
| 6 × 100 | 2/8 | S + PD | 2/8 | S + PD | ||
| 6 × 10−1 | 0/8 | PD | 0/8 | PD | ||
| Methanococcus maripaludis DSM 2067 | ||||||
| 2.11 × 106 | 8/8 | S | 9/9 | S | ||
| 2.11 × 105 | 8/8 | 9/9 | ||||
| 2.11 × 104 | 8/8 | 9/9 | ||||
| 2.11 × 103 | 8/8 | 9/9 | ||||
| 2.11 × 102 | 8/8 | 9/9 | ||||
| 2.11 × 101 | 8/8 | 2/8 | 9/9 | 3/9 | ||
| 6/8 | S + PD | 6/9 | S + PD | |||
| 2.11 × 100 | 2/8 | S + PD | 3/9 | S + PD | ||
| 2.11 × 10−1 | 0/8 | PD | 0/9 | PD | ||
| Methanomicrobium mobile DSM 1539 | ||||||
| 5.71 × 106 | 8/8 | S | 7/7 | S | ||
| 5.71 × 105 | 8/8 | 7/7 | ||||
| 5.71 × 104 | 9/9 | 7/7 | ||||
| 5.71 × 103 | 8/8 | 7/7 | ||||
| 5.71 × 102 | 9/9 | 1/9 | 7/7 | 4/7 | ||
| 8/9 | S + PD | 3/7 | S + PD | |||
| 5.71 × 101 | 1/8 | S + PD | 7/7 | S + PD | ||
| 5.71 × 100 | 0/8 | PD | 0/7 | PD | ||
| 5.71 × 10−1 | 0/8 | PD | 0/7 | PD | ||
| Estimated Plasmid/Genome Copy No. | Mlas/mcrA-Rev | mcrA_F3/mcrA-Rev | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Intra-Assay Variability | Inter-Assay Variability | Efficiency (%) | Intra-Assay Variability | Inter-Assay Variability | Efficiency (%) | |||||
| Mean Cq ± SD | CV% | Mean Cq ± SD | CV% | Mean Cq ± SD | CV% | Mean Cq ± SD | CV% | |||
| mcrA+ Plasmid | ||||||||||
| 6.24 × 106 | 13.32 ± 0.02 | 0.11 | 13.20 ± 0.14 | 1.03 | 91.69 | 14.94 ± 0.17 | 1.10 | 15.11 ± 0.03 | 0.20 | 90.92 |
| 6.24 × 105 | 17.09 ± 0.08 | 0.48 | 17.05 ± 0.35 | 2.05 | 19.16 ± 0.13 | 0.68 | 19.24 ± 0.19 | 0.97 | ||
| 6.24 × 104 | 21.08 ± 0.17 | 0.81 | 21.01 ± 0.47 | 2.24 | 23.48 ± 0.28 | 1.18 | 23.01 ± 0.4 | 1.76 | ||
| 6.24 × 103 | 24.72 ± 0.12 | 0.48 | 24.71 ± 0.24 | 0.96 | 27.32 ± 0.38 | 1.40 | 27.10 ± 0.29 | 1.05 | ||
| 6.24 × 102 | 28.26 ± 0.24 | 0.84 | 27.75 ± 0.55 | 1.97 | 30.91 ± 0.12 | 0.38 | 30.32 ± 0.8 | 2.64 | ||
| 6.24 × 101 | 30.58 ± 0.04 | 0.13 | 30.81 ± 0.57 | 1.85 | 33.77 ± 0.07 | 0.21 | 32.57 ± 0.77 | 2.36 | ||
| 6.24 × 100 | 31.56 * (1/3) | n/a | 31.34 * (2/3) ± 0.31 | 0.99 | n/a | 34.55 * (1/3) | n/a | 33.72 ± 1.05 | 3.11 | n/a |
| 6.24 × 10−1 | negative | negative | ||||||||
| Methanobrevibacter woesei DSM 11979 | ||||||||||
| 6 × 105 | 18.09 ± 0.21 | 1.14 | 18.45 ± 0.4 | 2.14 | 109.33 | 18.69 ± 0.34 | 1.81 | 19.62 ± 0.78 | 3.95 | 98.96 |
| 6 × 104 | 21.80 ± 0.12 | 0.56 | 21.63 ± 0.41 | 1.89 | 22.95 ±0.2 | 0.88 | 23.33 ± 0.51 | 2.18 | ||
| 6 × 103 | 25.38 ± 0.3 | 1.17 | 25.29 ± 0.4 | 1.57 | 26.89 ± 0.18 | 0.68 | 26.72 ± 0.08 | 0.29 | ||
| 6 × 102 | 28.69 ± 0.13 | 0.45 | 28.43 ± 0.33 | 1.15 | 30.87 ± 0.11 | 0.36 | 30.53 ± 0.24 | 0.79 | ||
| 6 × 101 | 30.48 ± 0.15 | 0.50 | 30.63 ± 0.05 | 0.17 | 32.88 ± 0.3 | 0.92 | 32.76 ± 0.25 | 0.77 | ||
| 6 × 100 | 30.82 * (2/3) ± 0.21 | 0.69 | 30.97 * (1/3) | n/a | n/a | negative | 33.90 * (2/3) ± 0.7 | 2.07 | n/a | |
| 6 × 10−1 | negative | negative | ||||||||
| Methanococcus maripaludis DSM 2067 | ||||||||||
| 2.11 × 106 | 14.41 ± 0.25 | 1.72 | 14.79 ± 0.05 | 0.35 | 103.18 | 16.09 ± 0.52 | 3.22 | 16.80 ± 0.28 | 1.64 | 90.84 |
| 2.11 × 105 | 18.07 ± 0.08 | 0.44 | 18.54 ± 0.04 | 0.24 | 20.30 ± 0.33 | 1.61 | 20.61 ± 0.15 | 0.71 | ||
| 2.11 × 104 | 22.53 ± 0.04 | 0.19 | 22.15 ± 0.3 | 1.37 | 24.26 ± 0.24 | 0.97 | 24.17 ± 0.31 | 1.29 | ||
| 2.11 × 103 | 25.98 ± 0.09 | 0.34 | 25.43 ± 0.52 | 2.06 | 28.14 ± 0.21 | 0.73 | 27.81 ± 0.63 | 2.27 | ||
| 2.11 × 102 | 29.21 ± 0.13 | 0.46 | 28.50 ± 0.61 | 2.14 | 31.81 ± 0.19 | 0.60 | 31.44 ± 0.44 | 1.40 | ||
| 2.11 × 101 | 30.84 ± 0.07 | 0.23 | 30.90 ± 0.17 | 0.55 | 34.87 ± 0.26 | 0.74 | 34.51 ± 0.81 | 2.36 | ||
| 2.11 × 100 | negative | 31.11 * (2/3) ± 0.06 | 0.18 | n/a | negative | 34.87 ± 0.59 | 1.68 | n/a | ||
| 2.11 × 10−1 | negative | negative | ||||||||
| Methanomicrobium mobile DSM 1539 | ||||||||||
| 5.71 × 106 | 18.47 ± 0.05 | 0.29 | 18.31 ± 0.21 | 1.12 | 104 | 20.19 ± 0.56 | 2.79 | 19.49 ± 0.78 | 4.01 | 97.67 |
| 5.71 × 105 | 22.22 ± 0.03 | 0.11 | 22.00 ± 0.22 | 1.00 | 24.22 ± 0.3 | 1.24 | 23.31 ± 0.79 | 3.38 | ||
| 5.71 × 104 | 25.85 ± 0.09 | 0.33 | 25.44 ± 0.42 | 1.63 | 28.32 ± 0.14 | 0.48 | 27.38 ± 0.74 | 2.69 | ||
| 5.71 × 103 | 29.34 ± 0.2 | 0.69 | 28.68 ± 0.41 | 1.42 | 32.46 ± 0.26 | 0.79 | 30.54 ± 0.82 | 2.67 | ||
| 5.71 × 102 | 30.65 ± 0.3 | 0.97 | 31.12 ± 0.16 | 0.52 | 35.21 ± 0.59 | 1.69 | 33.86 ± 1.07 | 3.17 | ||
| 5.71 × 101 | negative | 31.73 * (1/3) | n/a | n/a | 36.17 ± 0.79 | 2.18 | 36.19 ± 0.77 | 2.12 | ||
| 5.71 × 100 | negative | negative | ||||||||
| 5.71 × 10−1 | negative | negative | ||||||||
| DNA Template | Estimated Genome Copy No. Per Reaction or Amount * | Cq | |
|---|---|---|---|
| Mlas/mcrA-Rev | mcrA_F3/mcrA-Rev | ||
| Escherichia coli ATCC 8739 | 4.38 × 105 copies | 26.76 | 34.17 |
| Salmonella Typhimurium ATCC 14028 | 3.44 × 105 copies | 31.58 | 34.06 |
| Clostridium septicum ATCC 12464 | 4.48 × 105 copies | 31.19 | 32.98 |
| Lactobacillus sakei ATCC 15521 | 6.6 × 106 copies | 29.59 | 30.41 |
| Enterococcus avium ATCC 14025 | 5.86 × 106 copies | 26.14 | 32.41 |
| Enterococcus casseliflavus ATCC 700327 | 2.48 × 106 copies | 26.47 | 33.18 |
| Enterococcus raffinosus ATCC 49464 | 7.7 × 104 copies | 27.65 | 33.20 |
| Blautia obeum DSM 25238 | 2.08 × 107 copies | 28.11 | 34.28 |
| Ruminococcus gauvreauii DSM 19829 | 8.83 × 105 copies | 27.82 | 35.21 |
| Helicobacter cinaedi DSM 5359 | 1.71 × 107 copies | 24.63 | 36.36 |
| Desulfovibrio piger DSM 749 | 1.32 × 108 copies | 29.06 | 34.49 |
| Proteus sp. | 6.1 ng | 23.15 | 32.92 |
| Streptococcus sp. | 17 ng | 26.45 | 34.66 |
| Streptococcus sp. (beta-hemolytic) | 0.118 ng | 28.21 | 34.46 |
| Corynebacterium sp. | 1.15 ng | 30.43 | 36.49 |
| Pseudomonas aeruginosa | 70 ng | 26.35 | 32.14 |
| Pasteurella sp. | 0.377 ng | 31.43 | 37.80 |
| Klebsiella sp. | 28 ng | 26.23 | 32.45 |
| Staphylococcus sp. (coagulase-negative) | 3.28 ng | 26.98 | 36.10 |
| Porphyromonas sp. | 2.43 ng | 25.53 | 29.05 |
| Bacteroidetes bacterium | 1.13 ng | 27.18 | 36.13 |
| Sample | Mlas/mcrA-Rev | mcrA_F3/mcrA-Rev | ||
|---|---|---|---|---|
| Cq | Tm (°C) | Cq | Tm (°C) | |
| 1 | 24.82 | 77.84; 82.25 | 28.17 | 82.51 |
| 2 | 24.54 | 71.84 | 29.62 | 79.47; 83.69 |
| 3 | 24.50 | 77.94; 83.23 | 23.17 | 83.36 |
| 4 | 25.59 | 83.18; 88.91 | 27.98 | 82.55 |
| 5 | 25.16 | 83.43 | 27.63 | 83.59 |
| 6 | 28.19 | 74.85 | 31.14 | 80.32; 83.28 |
| 7 | 28.09 | 77.62 | 30.63 | 83.05 |
| 8 | negative | 75.82; 80.96 | 28.27 | 77.08; 83.67 |
| 9 | 27.20 | 83.07 | 25.34 | 83.13 |
| 10 | 25.57 | 79.63; 83.17 | 28.15 | 83.11 |
| 11 | 25.73 | 80.65 | 28.58 | 77.11; 83.31 |
| 12 | 27.93 | 77.41 | 30.53 | 83.11 |
| 13 | 27.67 | 75.94; 82.02 | 28.80 | 82.88 |
| 14 | 28.36 | 82.97 | 29.40 | 77.51; 82.87 |
| 15 | 30.45 | 79.71 | 31.22 | 76.09; 82.87 |
| 16 | 25.60 | 75.51; 81.37 | 29.85 | 82.98 |
| 17 | 26.47 | 76.13; 81.10 | 28.90 | 76.59; 81.80 |
| 18 | 28.14 | 75.72 | 29.86 | 77.92; 83.12 |
| 19 | 29.29 | 75.91; 83.01 | 29.93 | 83.12 |
| 20 | 26.96 | 74.38 | 28.48 | 82.88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cisek, A.A.; Bąk, I.; Cukrowska, B. Improved Quantitative Real-Time PCR Protocol for Detection and Quantification of Methanogenic Archaea in Stool Samples. Microorganisms 2023, 11, 660. https://doi.org/10.3390/microorganisms11030660
Cisek AA, Bąk I, Cukrowska B. Improved Quantitative Real-Time PCR Protocol for Detection and Quantification of Methanogenic Archaea in Stool Samples. Microorganisms. 2023; 11(3):660. https://doi.org/10.3390/microorganisms11030660
Chicago/Turabian StyleCisek, Agata Anna, Iwona Bąk, and Bożena Cukrowska. 2023. "Improved Quantitative Real-Time PCR Protocol for Detection and Quantification of Methanogenic Archaea in Stool Samples" Microorganisms 11, no. 3: 660. https://doi.org/10.3390/microorganisms11030660
APA StyleCisek, A. A., Bąk, I., & Cukrowska, B. (2023). Improved Quantitative Real-Time PCR Protocol for Detection and Quantification of Methanogenic Archaea in Stool Samples. Microorganisms, 11(3), 660. https://doi.org/10.3390/microorganisms11030660