

Dysregulation of Stress-Induced Translational Control by Porphyromonas gingivalis in Host Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Bacterial Strains and Culture
2.4. Bacterial Infection, Oxidative Stress Induction, and Cell Treatments
2.5. Isolation of Crude Preparations of P. gingivalis Outer Membrane Vesicles (OMVs)
2.6. Generation of P. gingivalis Conditioned Media and Gingipain Inhibition
2.7. Western Blotting
2.8. Puromycin Incorporation Assay
2.9. Immunocytochemistry
2.10. Statistical Analysis
3. Results
3.1. P. gingivalis Infection Heightens Translational Repression and Modulates Stress Granule Formation during Exogenous Stress
3.2. P. gingivalis Heightens Translational Repression Independently of eIF2α
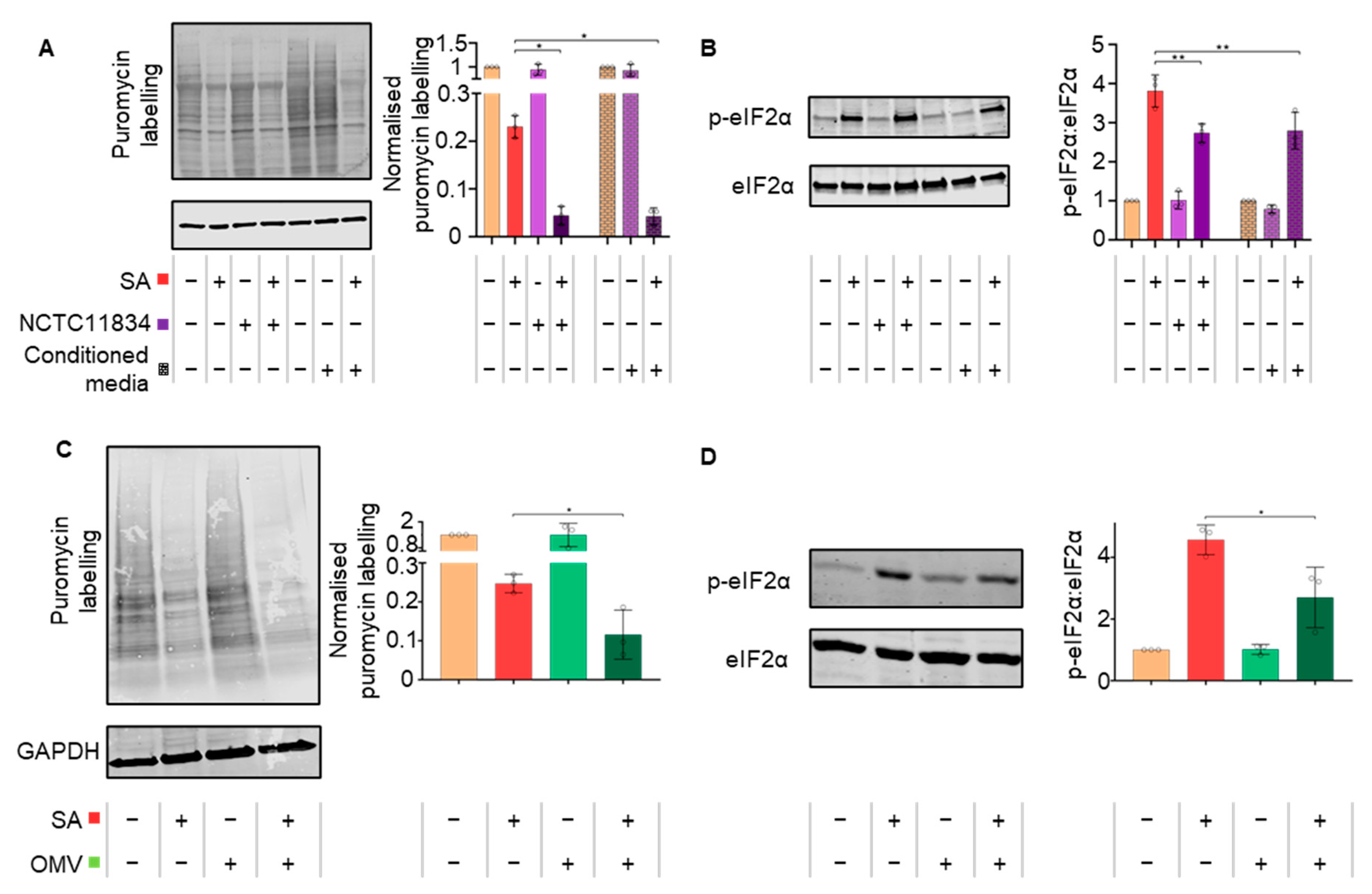
3.3. P. gingivalis Heightens Translational Repression via the Action of a Secretory Factor
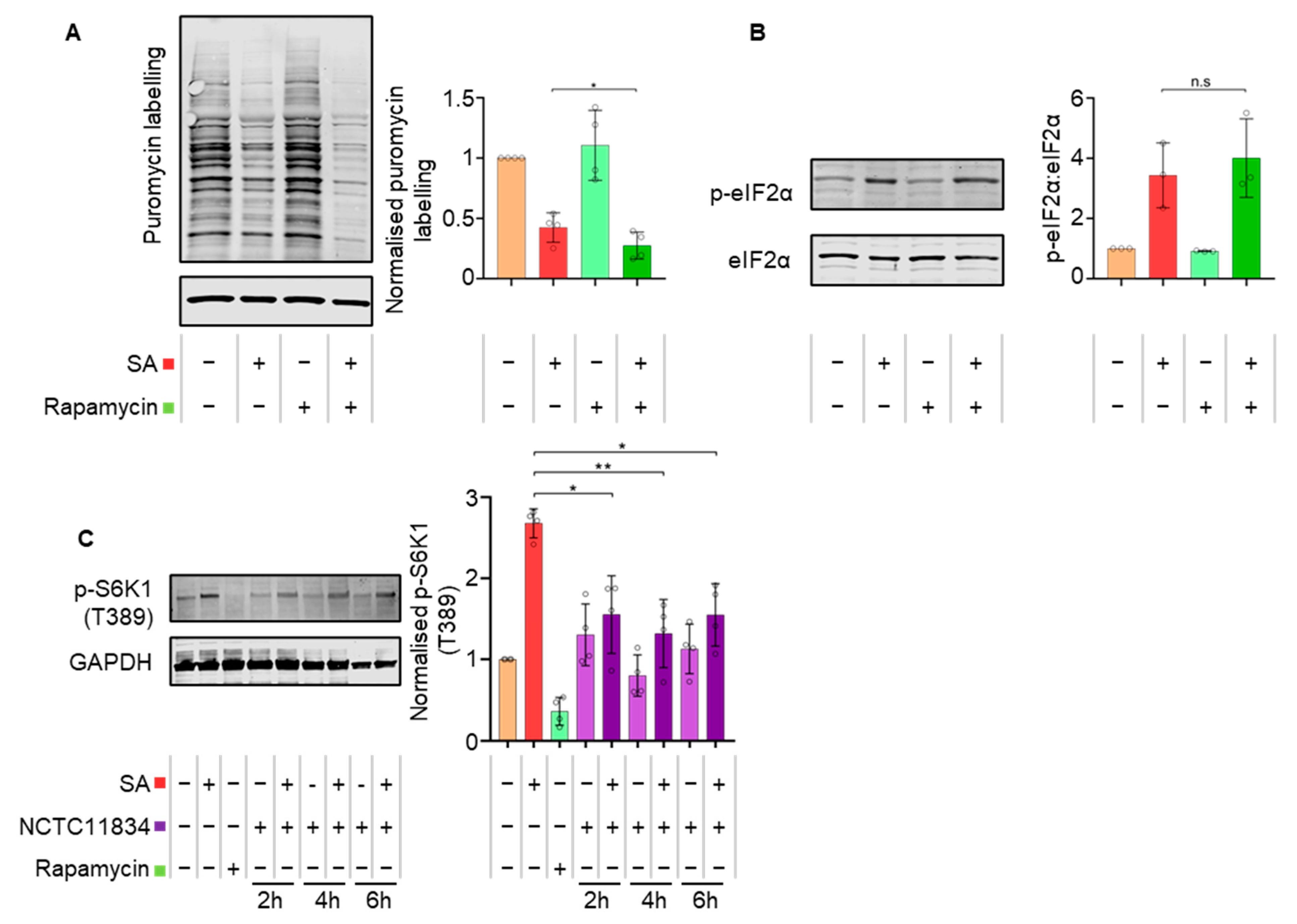
3.4. P. gingivalis Dysregulates mTOR Signaling during Stress
3.5. Secreted P. gingivalis Proteases, Gingipains, Mediate Heightened Translational Repression during Stress
3.6. P. gingivalis Infection Dampens Stress-Induced Tubulin Acetylation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marsh, P.D. Dental Plaque as a Microbial Biofilm. Caries Res. 2004, 38, 204–211. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Pihlstrom, B.L.; Michalowicz, B.S.; Johnson, N.W. Periodontal diseases. Lancet 2005, 366, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Tonetti, M.S.; Jepsen, S.; Jin, L.; Otomo-Corgel, J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. J. Clin. Periodontol. 2017, 44, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Kebschull, M.; Demmer, R.T.; Papapanou, P.N. “Gum Bug, Leave My Heart Alone!”—Epidemiologic and Mechanistic Evidence Linking Periodontal Infections and Atherosclerosis. J. Dent. Res. 2010, 89, 879–902. [Google Scholar] [CrossRef]
- Bingham, O.C.; Moni, O.M. Periodontal disease and rheumatoid arthritis: The evidence accumulates for complex pathobiologic interactions. Curr. Opin. Rheumatol. 2013, 25, 345–353. [Google Scholar] [CrossRef]
- Preshaw, P.; Alba, A.; Herrera, D.; Jepsen, S.; Konstantinidis, A.; Makrilakis, K.; Taylor, R. Periodontitis and diabetes: A two-way relationship. Diabetologia 2012, 55, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Gnanasekaran, J.; Binder Gallimidi, A.; Saba, E.; Pandi, K.; Eli Berchoer, L.; Hermano, E.; Angabo, S.; Makkawi, H.; Khashan, A.; Daoud, A.; et al. Intracellular Porphyromonas gingivalis Promotes the Tumorigenic Behavior of Pancreatic Carcinoma Cells. Cancers 2020, 12, 2331. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Adams, B.ü.; Nunes, J.M.; Page, M.J.; Roberts, T.; Carr, J.; Nell, T.A.; Kell, D.B.; Pretorius, E. Parkinson’s Disease: A Systemic Inflammatory Disease Accompanied by Bacterial Inflammagens. Front. Aging Neurosci. 2019, 11, 210. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Darveau, R.P.; Curtis, M.A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 2012, 10, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Lambris, J.D. Complement and dysbiosis in periodontal disease. Immunobiology 2012, 217, 1111–1116. [Google Scholar] [CrossRef]
- Grenier, D.; Chao, G.; Mcbride, B.C. Characterization of sodium dodecyl sulfate-stable Bacteroides gingivalis proteases by polyacrylamide gel electrophoresis. Infect. Immun. 1989, 57, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Collyer, C.A. Gingipains from Porphyromonas gingivalis—Complex domain structures confer diverse functions. Eur. J. Microbiol. Immunol. 2011, 1, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Stafford, P.; Higham, J.; Pinnock, A.; Murdoch, C.; Douglas, C.W.I.; Stafford, G.P.; Lambert, D.W. Gingipain-dependent degradation of mammalian target of rapamycin pathway proteins by the periodontal pathogen Porphyromonas gingivalis during invasion. Mol. Oral Microbiol. 2013, 28, 366–378. [Google Scholar] [CrossRef]
- Takahara, T.; Amemiya, Y.; Sugiyama, R.; Maki, M.; Shibata, H. Amino acid-dependent control of mTORC1 signaling: A variety of regulatory modes. J. Biomed. Sci. 2020, 27, 87. [Google Scholar] [CrossRef]
- Peake, J.; Suzuki, K. Neutrophil activation, antioxidant supplements and exercise-induced oxidative stress. Exerc. Immunol. Rev. 2004, 10, 129–141. [Google Scholar]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef]
- Knowles, A.; Campbell, S.; Cross, N.; Stafford, P. Bacterial manipulation of the Integrated Stress Response: A new perspective on infection. Front. Microbiol. 2021, 12, 645161. [Google Scholar] [CrossRef]
- Siekierka, J.; Mauser, L.; Ochoa, S. Mechanism of polypeptide chain initiation in eukaryotes and its control by phosphorylation of the alpha subunit of initiation factor 2. Proc. Natl. Acad. Sci. USA 1982, 79, 2537–2540. [Google Scholar] [CrossRef]
- Hinnebusch, A.G.; Lorsch, J.R. The mechanism of eukaryotic translation initiation: New insights and challenges. Cold Spring Harb. Perspect. Biol. 2012, 4, a011544. [Google Scholar] [CrossRef]
- Price, N.; Proud, C. The guanine nucleotide-exchange factor, eIF-2B. Biochimie 1994, 76, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.D.; Zhou, Y.; Mohammad-Qureshi, S.; Bennett, D.; Pavitt, G.D. eIF2B promotes eIF5 dissociation from eIF2*GDP to facilitate guanine nucleotide exchange for translation initiation. Genes Dev. 2013, 27, 2696–2707. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, A.G.; Panniers, R.; Henshaw, E.C. The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J. Biol. Chem. 1988, 263, 5526–5533. [Google Scholar] [CrossRef]
- Kenner, L.R.; Anand, A.A.; Nguyen, H.C.; Myasnikov, A.G.; Klose, C.J.; Mcgeever, L.A.; Tsai, J.C.; Miller-Vedam, L.; Walter, P.; Frost, A. eIF2B-catalyzed nucleotide exchange and phosphoregulation by the integrated stress response. Science 2019, 364, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Pause, A.; Belsham, G.J.; Anne-Claude Gingras Olivier, D.o.n.z.é.; Tai-An Lin Lawrence, J.C.; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5’-cap function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Heberle, A.M.; Prentzell, M.T.; van Eunen, K.; Bakker, B.M.; Grellscheid, S.N.; Thedieck, K. Molecular mechanisms of mTOR regulation by stress. Mol. Cell. Oncol. 2015, 2, e970489. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. Stressful initiations. J. Cell. Sci. 2002, 115, 3227–3234. [Google Scholar] [CrossRef]
- Kedersha, N.; Cho, M.R.; Li, W.; Yacono, P.W.; Chen, S.; Gilks, N.; Golan, D.E.; Anderson, P. Dynamic Shuttling of Tia-1 Accompanies the Recruitment of mRNA to Mammalian Stress Granules. J. Cell Biol. 2000, 151, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Loschi, M.; Leishman, C.C.; Berardone, N.; Boccaccio, G.L. Dynein and kinesin regulate stress-granule and P-body dynamics. J. Cell. Sci. 2009, 122, 3973–3982. [Google Scholar] [CrossRef] [PubMed]
- Rabouw, H.H.; Visser, L.J.; Passchier, T.C.; Langereis, M.A.; Liu, F.; Giansanti, P.; van Vliet, A.L.W.; Dekker, J.G.; van der Grein, S.G.; Saucedo, J.G.; et al. Inhibition of the integrated stress response by viral proteins that block p-eIF2–eIF2B association. Nat. Microbiol. 2020, 5, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Nour, M.; Carneiro, L.A.M.; Downey, J.; Tsalikis, J.; Outlioua, A.; Prescott, D.; Da Costa, L.S.; Hovingh, E.S.; Farahvash, A.; Gaudet, R.G.; et al. The heme-regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science 2019, 365, eaaw4144. [Google Scholar] [CrossRef]
- Tattoli, I.; Sorbara, M.T.; Vuckovic, D.; Ling, A.; Soares, F.; Carneiro, L.A.M.; Yang, C.; Emili, A.; Philpott, D.J.; Girardin, S.E. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 2012, 11, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Van’t Wout, E.F.A.; van Schadewijk, A.; van Boxtel, R.; Dalton, L.E.; Clarke, H.J.; Tommassen, J.; Marciniak, S.J.; Hiemstra, P.S.; Parsek, M.R. Virulence Factors of Pseudomonas aeruginosa Induce Both the Unfolded Protein and Integrated Stress Responses in Airway Epithelial Cells. PLoS Pathog. 2015, 11, e1004946. [Google Scholar] [CrossRef] [PubMed]
- Tsutsuki, H.; Yahiro, K.; Ogura, K.; Ichimura, K.; Iyoda, S.; Ohnishi, M.; Nagasawa, S.; Seto, K.; Moss, J.; Noda, M. Subtilase cytotoxin produced by locus of enterocyte effacement-negative Shiga-toxigenic Escherichia coli induces stress granule formation. Cell. Microbiol. 2016, 18, 1024–1040. [Google Scholar] [CrossRef]
- Velásquez, F.; Marín-Rojas, J.; Soto-Rifo, R.; Torres, A.; Del Canto, F.; Valiente-Echeverría, F. Escherichia coli HS and Enterotoxigenic Escherichia coli Hinder Stress Granule Assembly. Microorganisms 2020, 9, 17. [Google Scholar] [CrossRef]
- Vonaesch, P.; Campbell-Valois, F.; Dufour, A.; Sansonetti, P.J.; Schnupf, P. Shigella flexneri modulates stress granule composition and inhibits stress granule aggregation. Cell. Microbiol. 2016, 18, 982–997. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Inoue, T.; Naito, M.; Nakayama, K.; Ohara, N. Attenuation of the Phosphatidylinositol 3-Kinase/Akt Signaling Pathway by Porphyromonas gingivalis Gingipains RgpA, RgpB, and Kgp. J. Biol. Chem. 2015, 290, 5190–5202. [Google Scholar] [CrossRef]
- Hirasawa, M.; Kurita-Ochiai, T. Porphyromonas gingivalis Induces Apoptosis and Autophagy via ER Stress in Human Umbilical Vein Endothelial Cells. Mediat. Inflamm. 2018, 2018, 1967506. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell. 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Aduse-Opoku, J.; Davies, N.N.; Gallagher, A.; Hashim, A.; Evans, H.E.A.; Rangarajan, M.; Slaney, J.M.; Curtis, M.A. Generation of Lys-gingipain protease activity in Porphyromonas gingivalis W50 is independent of Arg-gingipain protease activities. Microbiology 2000, 146, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Ho, M.; Liu, B.; Hildreth, J.; Dash, C.; Goodwin, J.S.; Balasubramaniam, M.; Chen, C.; Xie, H. Role of Porphyromonas gingivalis outer membrane vesicles in oral mucosal transmission of HIV. Sci. Rep. 2018, 8, 8812. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, R.E.; Varanda, B.A.; Ashe, M.P.; Allen, K.E.; Campbell, S.G. Cellular eIF2B subunit localization: Implications for the integrated stress response and its control by small molecule drugs. Mol. Biol. Cell. 2019, 30, 942–958. [Google Scholar] [CrossRef] [PubMed]
- Aulas, A.; Fay, M.M.; Lyons, S.M.; Achorn, C.A.; Kedersha, N.; Anderson, P.; Ivanov, P. Stress-specific differences in assembly and composition of stress granules and related foci. J. Cell. Sci. 2017, 130, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. eLife 2015, 4, e05033. [Google Scholar] [CrossRef]
- Nandagopal, N.; Roux, P.P. Regulation of global and specific mRNA translation by the mTOR signaling pathway. Translation 2015, 3, e983402. [Google Scholar] [CrossRef]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct stages in stress granule assembly and disassembly. eLife 2016, 5, e18413. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Pavitt, G.D.; Ron, D. New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harb. Perspect. Biol. 2012, 4, a012278. [Google Scholar] [CrossRef] [PubMed]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef]
- Lee, P.; Ho, I.; Lee, T. Oxidative Stress Mediates Sodium Arsenite-Induced Expression of Heme Oxygenase-1, Monocyte Chemoattractant Protein-1, and Interleukin-6 in Vascular Smooth Muscle Cells. Toxicol. Sci. 2005, 85, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Rabouw, H.H.; Langereis, M.A.; Anand, A.A.; Visser, L.J.; de Groot, R.J.; Walter, P.; van Kuppeveld, F.J.M. Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2097–2102. [Google Scholar] [CrossRef] [PubMed]
- Brostrom, C.O.; Prostko, C.R.; Kaufman, R.J.; Brostrom, M.A. Inhibition of translational initiation by activators of the glucose-regulated stress protein and heat shock protein stress response systems. Role of the interferon-inducible double-stranded RNA-activated eukaryotic initiation factor 2alpha kinase. J. Biol. Chem. 1996, 271, 24995. [Google Scholar] [CrossRef]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-Binding Proteins Tia-1 and Tiar Link the Phosphorylation of Eif-2α to the Assembly of Mammalian Stress Granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef]
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and immune pathways in the pathogenesis of periodontal disease: Inflammatory and immune pathways in periodontal disease. Periodontology 2000 2014, 64, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Henry, L.G.; McKenzie, R.M.E.; Robles, A.; Fletcher, H.M. Oxidative stress resistance in Porphyromonas gingivalis. Future Microbiol. 2012, 7, 497–512. [Google Scholar] [CrossRef]
- Lamont, R.J.; Chan, A.; Belton, C.M.; Izutsu, K.T.; Vasel, D.; Weinberg, A. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect. Immun. 1995, 63, 3878–3885. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, H.; Duan, X.; Jotwani, R.; Vuddaraju, H.; Liang, S.; Scott, D.A.; Lamont, R.J. Porphyromonas gingivalis-induced reactive oxygen species activate JAK2 and regulate production of inflammatory cytokines through c-Jun. Infect. Immun. 2014, 82, 4118–4126. [Google Scholar] [CrossRef]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö.P. gingivalis-Nucleoside-diphosphate-kinase Inhibits ATP-Induced Reactive-Oxygen-Species via P2X7 Receptor/NADPH-Oxidase Signaling and Contributes to Persistence. Cell. Microbiol. 2013, 15, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.W.; Birss, A.J.; Silver, J. The periodontal pathogen Porphyromonas gingivalis harnesses the chemistry of the μ-oxo bishaem of iron protoporphyrin IX to protect against hydrogen peroxide. FEMS Microbiol. Lett. 2000, 183, 159–164. [Google Scholar] [CrossRef]
- Johnson, N.A.; McKenzie, R.; Mclean, L.; Sowers, L.C.; Fletcher, H.M. 8-Oxo-7,8-Dihydroguanine Is Removed by a Nucleotide Excision Repair-Like Mechanism in Porphyromonas gingivalis W83. J. Bacteriol. 2004, 186, 7697–7703. [Google Scholar] [CrossRef]
- Yancy, S.L.; Shelden, E.A.; Gilmont, R.R.; Welsh, M.J. Sodium Arsenite Exposure Alters Cell Migration, Focal Adhesion Localization and Decreases Tyrosine Phosphorylation of Focal Adhesion Kinase in H9C2 Myoblasts. Toxicol. Sci. 2005, 84, 278–286. [Google Scholar] [CrossRef]
- McEwen, E.; Kedersha, N.; Song, B.; Scheuner, D.; Gilks, N.; Han, A.; Chen, J.; Anderson, P.; Kaufman, R.J. Heme-regulated Inhibitor Kinase-mediated Phosphorylation of Eukaryotic Translation Initiation Factor 2 Inhibits Translation, Induces Stress Granule Formation, and Mediates Survival upon Arsenite Exposure. J. Biol. Chem. 2005, 280, 16925–16933. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Y.; Markmiller, S.; Vu, A.Q.; Javaherian, A.; Dowdle, W.E.; Jolivet, P.; Bushway, P.J.; Castello, N.A.; Baral, A.; Chan, M.Y.; et al. Small-Molecule Modulation of TDP-43 Recruitment to Stress Granules Prevents Persistent TDP-43 Accumulation in ALS/FTD. Neuron 2019, 103, 802–819.e11. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.; Raught, B.; Sonenberg, N. eIF4 Initiation Factors: Effectors of mRNA Recruitment to Ribosomes and Regulators of Translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef] [PubMed]
- Sévigny, M.; Bourdeau Julien, I.; Venkatasubramani, J.P.; Hui, J.B.; Dutchak, P.A.; Sephton, C.F. FUS contributes to mTOR-dependent inhibition of translation. J. Biol. Chem. 2020, 295, 18459–18473. [Google Scholar] [CrossRef] [PubMed]
- Larsson, O.; Morita, M.; Topisirovic, I.; Alain, T.; Blouin, M.; Pollak, M.; Sonenberg, N. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc. Natl. Acad. Sci. USA 2012, 109, 8977–8982. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, X.; Wu, S.; Lu, J.; Zheng, M.; Wang, Y.; Zhou, H.; Zhang, H.; Han, J. Phosphorylation of Raptor by p38β Participates in Arsenite-induced Mammalian Target of Rapamycin Complex 1 (mTORC1) Activation. J. Biol. Chem. 2011, 286, 31501–31511. [Google Scholar] [CrossRef] [PubMed]
- Nadezhdina, E.S.; Lomakin, A.J.; Shpilman, A.A.; Chudinova, E.M.; Ivanov, P.A. Microtubules govern stress granule mobility and dynamics. BBA Mol. Cell Res. 2010, 1803, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Kinane, J.A.; Benakanakere, M.R.; Zhao, J.; Hosur, K.B.; Kinane, D.F. Porphyromonas gingivalis influences actin degradation within epithelial cells during invasion and apoptosis. Cell. Microbiol. 2012, 14, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Magiera, M.M.; Janke, C. Post-translational modifications of tubulin. Curr. Biol. 2014, 24, R351–R354. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jayabal, S.; Ghorbani, M.; Legault, L.; Mcgraw, S.; Watt, A.J.; Yang, X. ATAT1 regulates forebrain development and stress-induced tubulin hyperacetylation. Cell. Mol. Life Sci. 2019, 76, 3621–3640. [Google Scholar] [CrossRef]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef]
- Cai, D.; McEwen, D.P.; Martens, J.R.; Meyhofer, E.; Verhey, K.J.; Schliwa, M. Single Molecule Imaging Reveals Differences in Microtubule Track Selection Between Kinesin Motors. PLoS Biol. 2009, 7, e1000216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. Embo J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef]
- Kwon, S.; Zhang, Y.; Matthias, P. The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev. 2007, 21, 3381–3394. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; Zhang, S.; et al. The role of host eIF2α in viral infection. Virol. J. 2020, 17, 112. [Google Scholar] [CrossRef]
- Schoof, M.; Boone, M.; Wang, L.; Lawrence, R.; Frost, A.; Walter, P. eIF2B Conformation and Assembly State Regulate the Integrated Stress Response. FASEB J. 2021, 35. [Google Scholar] [CrossRef]
- Potempa, J.; Travis, J. Porphyromonas gingivalis proteinases in periodontitis, a review. Acta Biochim. Pol. 1996, 43, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, H.; Duncan, M.J. Clathrin-dependent entry of a gingipain adhesin peptide and Porphyromonas gingivalis into host cells. Cell. Microbiol. 2008, 10, 2538–2552. [Google Scholar] [CrossRef] [PubMed]
- Mantri, C.K.; Chen, C.; Dong, X.; Goodwin, J.S.; Pratap, S.; Paromov, V.; Xie, H. Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiologyopen 2015, 4, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Wang, T.; Taub, F.; Park, Y.; Capestany, C.A.; Lamont, R.J.; Hackett, M. Quantitative proteomics of intracellular Porphyromonas gingivalis. Proteomics 2007, 7, 4323–4337. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Vaklavas, C.; Blume, S.W.; Grizzle, W.E. Translational Dysregulation in Cancer: Molecular Insights and Potential Clinical Applications in Biomarker Development. Front. Oncol. 2017, 7, 158. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Darzi, Y.; Tawaratsumida, K.; Marchesan, J.T.; Hasegawa, M.; Moon, H.; Chen, G.Y.; Núñez, G.; Giannobile, W.V.; Raes, J.; et al. Induction of Bone Loss by Pathobiont-Mediated Nod1 Signaling in the Oral Cavity. Cell Host Microbe 2013, 13, 595–601. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knowles, A.A.; Campbell, S.G.; Cross, N.A.; Stafford, P. Dysregulation of Stress-Induced Translational Control by Porphyromonas gingivalis in Host Cells. Microorganisms 2023, 11, 606. https://doi.org/10.3390/microorganisms11030606
Knowles AA, Campbell SG, Cross NA, Stafford P. Dysregulation of Stress-Induced Translational Control by Porphyromonas gingivalis in Host Cells. Microorganisms. 2023; 11(3):606. https://doi.org/10.3390/microorganisms11030606
Chicago/Turabian StyleKnowles, Alex A., Susan G. Campbell, Neil A. Cross, and Prachi Stafford. 2023. "Dysregulation of Stress-Induced Translational Control by Porphyromonas gingivalis in Host Cells" Microorganisms 11, no. 3: 606. https://doi.org/10.3390/microorganisms11030606
APA StyleKnowles, A. A., Campbell, S. G., Cross, N. A., & Stafford, P. (2023). Dysregulation of Stress-Induced Translational Control by Porphyromonas gingivalis in Host Cells. Microorganisms, 11(3), 606. https://doi.org/10.3390/microorganisms11030606