
Comparing the Efficacy of MALDI-TOF MS and Sequencing-Based Identification Techniques (Sanger and NGS) to Monitor the Microbial Community of Irrigation Water
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolation
2.2. MALDI-TOF MS Identification
2.3. DNA Extraction and Sanger Sequencing of Waterborne Isolates
2.4. DNA Extraction and Next-Generation Sequencing of Irrigation Water Samples
2.5. Bioinformatics and Data Processing of Next-Generation Sequencing Data
2.6. Statistical Methods
3. Results
3.1. Results of MALDI-TOF MS Identification and Sanger Sequencing of Isolates
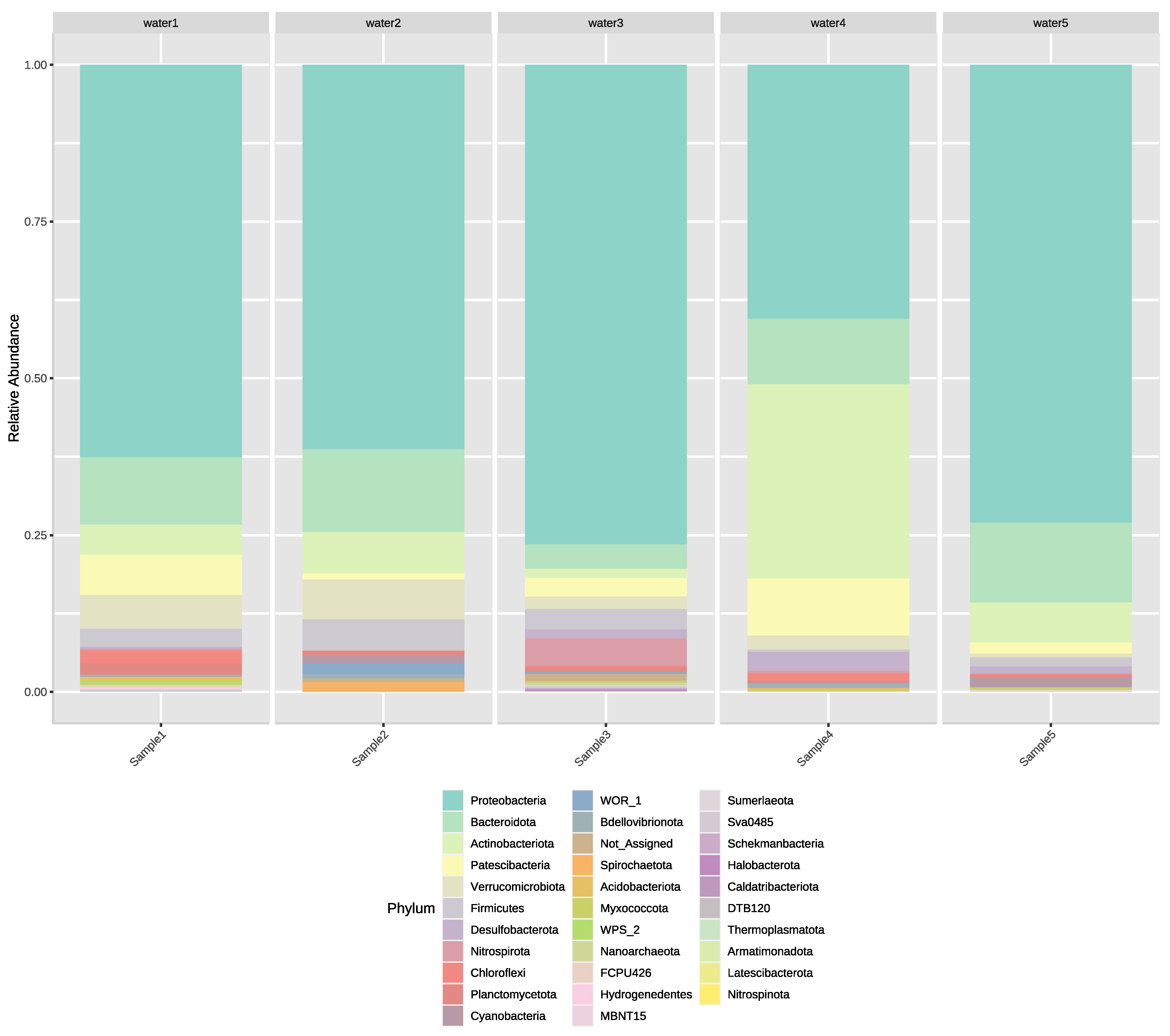
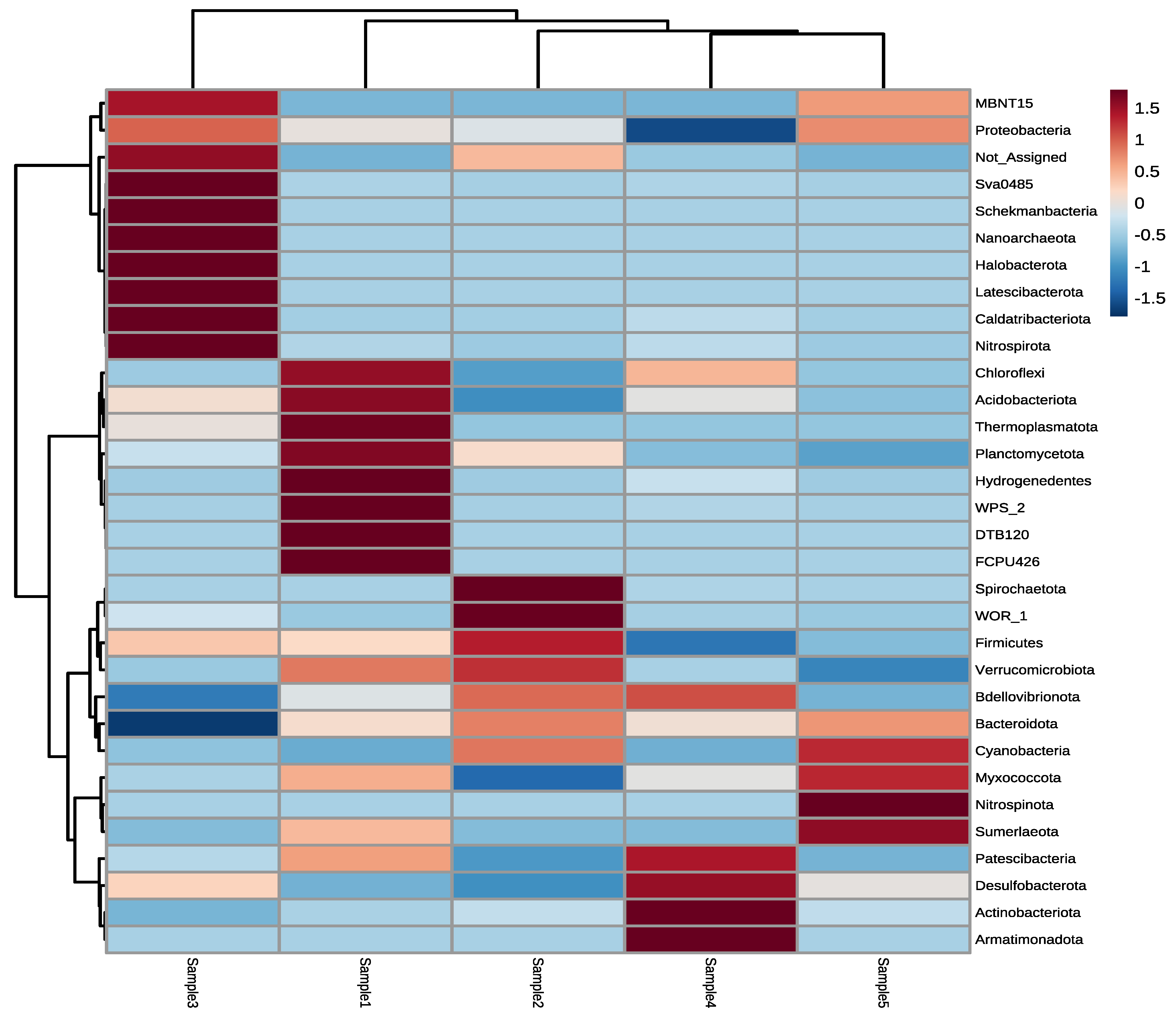
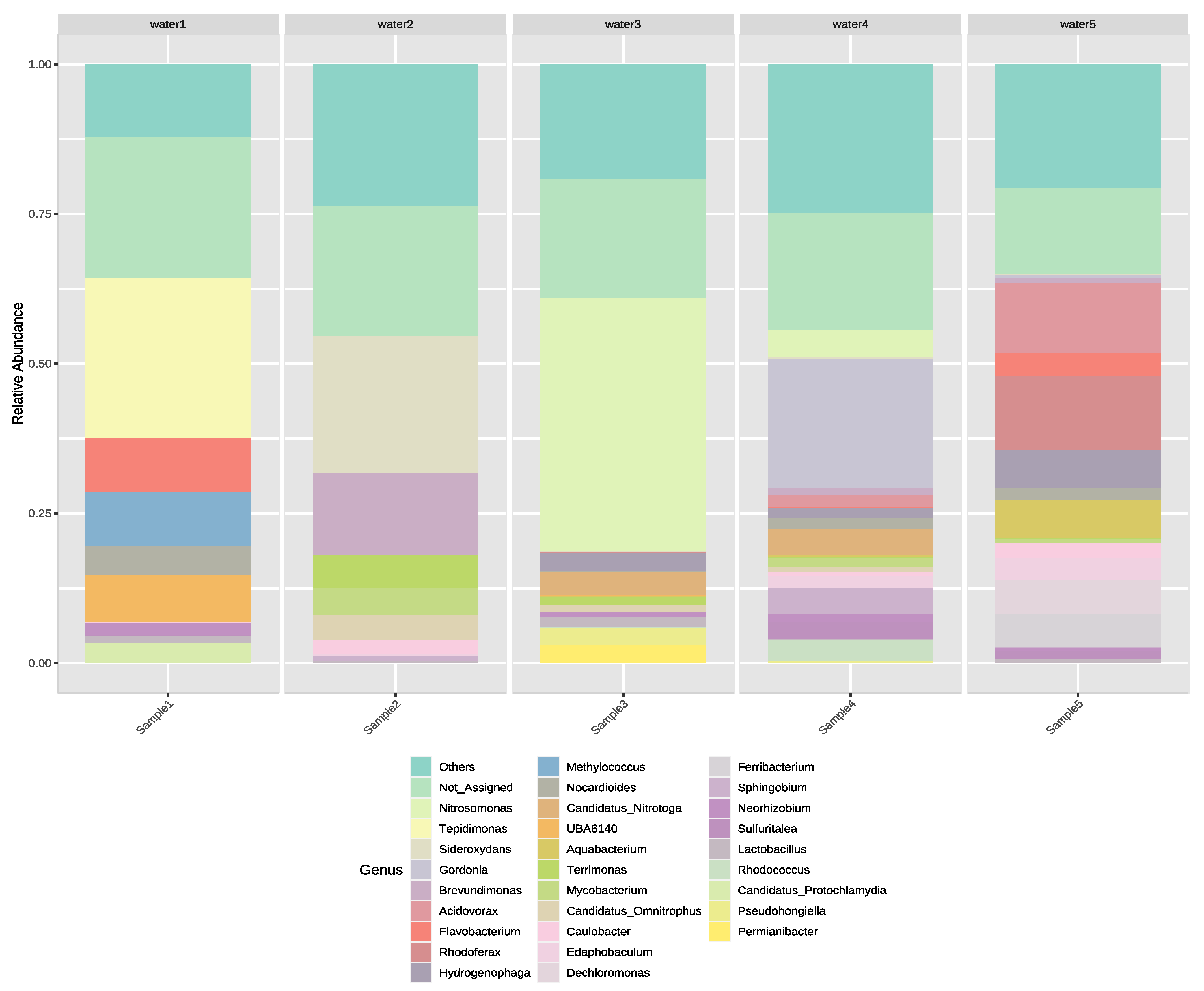
3.2. Next-Generation Sequencing of Irrigation Water
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gu, G.; Luo, Z.; Cevallos-Cevallos, J.M.; Adams, P.; Vellidis, G.; Wright, A.; van Bruggen, A.H. Factors affecting the occurrence of Escherichia coli O157 contamination in irrigation ponds on produce farms in the Suwannee River Watershed. Can. J. Microbiol. 2013, 59, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Uyttendaele, M.; Jaykus, L.-A.; Amoah, P.; Chiodini, A.; Cunliffe, D.; Jacxsens, L.; Holvoet, K.; Korsten, L.; Lau, M.; McClure, P.; et al. Microbial Hazards in Irrigation Water: Standards, Norms, and Testing to Manage Use of Water in Fresh Produce Primary Production. Compr. Rev. Food Sci. 2015, 14, 336–356. [Google Scholar] [CrossRef]
- Cevallos-Cevallos, J.M.; Gu, G.; Richardson, S.M.; Hu, J.; van Bruggen, A.H. Survival of Salmonella enterica Typhimurium in water amended with manure. J. Food Prot. 2014, 77, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Falardeau, J.; Johnson, R.P.; Pagotto, F.; Wang, S. Occurrence, characterization, and potential predictors of verotoxigenic Escherichia coli, Listeria monocytogenes, and Salmonella in surface water used for produce irrigation in the Lower Mainland of British Columbia, Canada. PloS ONE 2017, 27, 9. [Google Scholar] [CrossRef]
- European Food Safety Authority, European Centre for Disease Prevention and Control. The European Union One Health 2018 Zoonoses Report. EFSA J. 2019, 17, e05926. [Google Scholar] [CrossRef]
- Turner, K.; Moua, C.N.; Hajmeer, M.; Barnes, A.; Needham, M. Overview of Leafy Greens–Related Food Safety Incidents with a California Link: 1996 to 2016. J. Food Prot. 2019, 82, 405–414. [Google Scholar] [CrossRef]
- European Food Safety Authority, European Centre for Disease Prevention and Control. The European Union One Health 2019 Zoonoses Report. EFSA J. 2021, 19, e06406. [Google Scholar] [CrossRef]
- Thanner, S.; Drissner, D.; Walsh, F. Antimicrobial Resistance in Agriculture. mBio 2016, 7, 2. [Google Scholar] [CrossRef]
- Cerqueira, F.; Matamoros, V.; Bayona, J.; Elsinga, G.; Hornstra, L.M.; Piña, B. Distribution of antibiotic resistance genes in soils and crops. A field study in legume plants (Vicia faba L.) grown under different watering regimes. Environ. Res. 2019, 170, 16–25. [Google Scholar] [CrossRef]
- Azanu, D.; Styrishave, B.; Darko, G.; Weisser, J.J.; Abaidoo, R.C. Occurrence and risk assessment of antibiotics in water and lettuce in Ghana. Sci. Total Environ. 2018, 622–623, 293–305. [Google Scholar] [CrossRef]
- Pan, M.; Chu, L.M. Occurrence of antibiotics and antibiotic resistance genes in soils from wastewater irrigation areas in the Pearl River Delta region, southern China. Sci. Total Environ. 2018, 624, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chan, E.W.-C.; Miaomiao, X.; Liangwei, Y.; Ning, D.; Sheng, C. Widespread distribution of mcr-1-bearing bacteria in the ecosystem, 2015 to 2016. Euro Surveill. 2017, 22, 39. [Google Scholar] [CrossRef]
- Czekalski, N.; Berthold, T.; Caucci, S.; Egli, A.; Bürgmann, H. Increased levels of multiresistant bacteria and resistance genes after wastewater treatment and their dissemination into Lake Geneva, Switzerland. Front. Microbiol. 2012, 3, 106. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, J.R.; Ferreira, C.R.; Sanvido, G.B.; Kostrzewa, M.; Maier, T.; Wegemann, B.; Böttcher, V.; Eberlin, M.N.; dos Santos, M.V. Short communication: Identification of subclinical cow mastitis pathogens in milk by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Int. J. Dairy Sci. 2010, 93, 5661–5667. [Google Scholar] [CrossRef] [PubMed]
- Böhme, K.; Fernández-No, I.C.; Barros-Velázquez, J.; Gallardo, J.M.; Cañas, B.; Calo-Mata, P. Rapid species identification of seafood spoilage and pathogenic Gram-positive bacteria by MALDI-TOF mass fingerprinting. Electrophoresis 2011, 32, 2951–2965. [Google Scholar] [CrossRef]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef]
- Uchida-Fuji, E.; Niwa, H.; Kinoshita, Y.; Nukada, T. Matrix-Assisted Laser Desorption Ionization–Time of Flight Mass Spectrometry (MALDI-TOF MS) for Identification of Bacterial Isolates from Horses. J. Equine Vet. Sci. 2020, 95, 103276. [Google Scholar] [CrossRef]
- Avanzi, I.R.; Gracioso, L.H.; dos Passos Galluzzi Baltazar, M.; Karolski, B.; Perpetuo, E.A.; Nascimento, C.A.O. Rapid bacteria identification from environmental mining samples using MALDI-TOF MS analysis. Environ. Sci. Pollut. Res. 2017, 24, 3717–3726. [Google Scholar] [CrossRef]
- Pandey, A.; Jain, R.; Sharma, A.; Dhakar, K.; Kaira, G.S.; Rahi, P.; Dhyani, A.; Pandey, N.; Adhikari, P.; Shouche, Y.S. 16S rRNA gene sequencing and MALDI-TOF mass spectrometry based comparative assessment and bioprospection of psychrotolerant bacteria isolated from high altitudes under mountain ecosystem. SN Appl. Sci. 2019, 1, 278. [Google Scholar] [CrossRef]
- Strejcek, M.; Smrhova, T.; Junkova, P.; Uhlik, O. Whole-Cell MALDI-TOF MS Versus 16S rRNA Gene Analysis for Identification and Dereplication of Recurrent Bacterial Isolates. Front. Microbiol. 2018, 9, 1294. [Google Scholar] [CrossRef]
- El-Nemr, I.M.; Mushtaha, M.; Sundararaju, S.; Fontejon, C.; Suleiman, M.; Tang, P.; Goktepe, I.; Hasan, M.R. Application of MALDI Biotyper System for Rapid Identification of Bacteria Isolated from a Fresh Produce Market. Curr. Microbiol. 2019, 76, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Locey, K.J.; Lennon, J.T. Scaling laws predict global microbial diversity. Proc. Natl. Acad. Sci. USA. 2016, 113, 5970–5975. [Google Scholar] [CrossRef]
- Saunders, A.M.; Albertsen, M.; Vollertsen, J.; Nielsen, P.H. The activated sludge ecosystem contains a core community of abundant organisms. ISME J. 2016, 1, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Iliev, I.; Yahubyan, G.; Marhova, M.; Apostolova, E.; Gozmanova, M.; Gecheva, G.; Kostadinova, S.; Ivanova, A.; Baev, V. Metagenomic profiling of the microbial freshwater communities in two Bulgarian reservoirs. J. Basic Microbiol. 2017, 57, 669–679. [Google Scholar] [CrossRef]
- Jesser, K.J.; Noble, R.T. Vibrio Ecology in the Neuse River Estuary, North Carolina, Characterized by Next-Generation Amplicon Sequencing of the Gene Encoding Heat Shock Protein 60 (hsp60). Appl. Environ. Microbiol. 2018, 84, 13. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Kong, X.; Cui, B.; Jin, S.; Xie, Y.; Wang, X.; Deng, Y. Bacterial communities and potential waterborne pathogens within the typical urban surface waters. Sci. Rep. 2018, 8, 13368. [Google Scholar] [CrossRef]
- Wu, H.; Li, Y.; Zhang, W.; Wang, C.; Wang, P.; Niu, L.; Du, J.; Gao, Y. Bacterial community composition and function shift with the aggravation of water quality in a heavily polluted river. J. Environ. Manag. 2019, 237, 433–441. [Google Scholar] [CrossRef]
- Pinel, I.S.M.; Moed, D.H.; Vrouwenvelder, J.S.; van Loosdrecht, M.C.M. Bacterial community dynamics and disinfection impact in cooling water systems. Water Res. 2020, 172, 115505. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef]
- Kim, M.; Oh, H.S.; Park, S.C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351, Erratum in Int. J. Syst. Evol. Microbiol. 2014, 64, 1825. [Google Scholar] [CrossRef]
- Schloss, P.D.; Handelsman, J. Metagenomics for studying unculturable microorganisms: Cutting the Gordian knot. Genome Biol. 2005, 6, 229. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.r-project.org (accessed on 10 October 2022).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst—A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, 80–188. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Prot. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Kitzinger, K.; Koch, H.; Lücker, S.; Sedlacek, C.J.; Herbold, C.; Schwarz, J.; Daebeler, A.; Mueller, A.J.; Lukumbuzya, M.; Romano, S.; et al. Characterization of the First “Candidatus Nitrotoga” Isolate Reveals Metabolic Versatility and Separate Evolution of Widespread Nitrite-Oxidizing Bacteria. mBio 2018, 9, e01186-18. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wu, Y.-H.; Jian, S.-L.; Wang, C.-H.; Wu, M.; Cheng, L.; Xu, X.-W. Pseudohongiella nitratireducens sp. nov., isolated from seawater, and emended description of the genus Pseudohongiella. Int. J. Syst. Evol. 2016, 66, 12. [Google Scholar] [CrossRef]
- Scott, J.S.; Sterling, S.A.; To, H.; Seals, S.R.; Jones, A.E. Diagnostic performance of matrix-assisted laser desorption ionization time-of-flight mass spectrometry in blood bacterial infections: A systematic review and meta-analysis. Infect. Dis. 2016, 48, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Martiny, D.; Busson, L.; Wybo, I.; El Haj, R.A.; Dediste, A.; Vandenberg, O. Comparison of the Microflex LT and Vitek MS systems for routine identification of bacteria by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Clin. Microbiol. 2012, 50, 1313–1325. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.T.; Qin, T.T.; Bi, R.R.; Kang, H.Q.; Ma, P.; Gu, B. Performance of the matrix-assisted laser desorption ionization time-of flight mass spectrometry system for rapid identification of streptococci: A review. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 1005–1012. [Google Scholar] [CrossRef]
- Christner, M.; Rohde, H.; Wolters, M.; Sobottka, I.; Wegscheider, K.; Aepfelbacher, M. Rapid identification of bacteria from positive blood culture bottles by use of matrix-assisted laser desorption-ionization time of flight mass spectrometry fingerprinting. J. Clin. Microbiol. 2010, 48, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.; Sánchez-Juanes, F.; González-Avila, M.; Cembrero-Fuciños, D.; Herrero-Hernández, A.; González-Buitrago, J.M.; Muñoz-Bellido, J.L. Direct identification of urinary tract pathogens from urine samples by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Clin. Microbiol. 2010, 48, 2110–2115. [Google Scholar] [CrossRef]
- Mauchline, T.H.; Malone, J.G. Life in earth—The root microbiome to the rescue? Curr. Opin. Microbiol. 2017, 37, 23–28. [Google Scholar] [CrossRef]
- Zancarini, A.; Echenique-Subiabre, I.; Debroas, D.; Taïb, N.; Quiblier, C.; Humbert, J.F. Deciphering biodiversity and interactions between bacteria and microeukaryotes within epilithic biofilms from the Loue River, France. Sci. Rep. 2017, 7, 4344. [Google Scholar] [CrossRef]
- Kopcakova, A.; Stramova, Z.; Kvasnova, S.; Godany, A.; Perhacova, Z.; Pristas, P. Need for database extension for reliable identification of bacteria from extreme environments using MALDI TOF mass spectrometry. Chem. Pap. 2014, 68, 1435–1442. [Google Scholar] [CrossRef]
- Suzuki, Y.; Niina, K.; Matsuwaki, T.; Nukazawa, K.; Iguchi, A. (2018) Bacterial flora analysis of coliforms in sewage, river water, and ground water using MALDI-TOF mass spectrometry. J. Environ. Sci. Health A. 2018, 53, 160–173. [Google Scholar] [CrossRef]
- Böhme, K.; Fernández-No, I.C.; Pazos, M.; Gallardo, J.M.; Barros-Velázquez, J.; Canas, B.; Calo-Mata, P. Identification and classification of seafood-borne pathogenic and spoilage bacteria: 16S rRNA sequencing versus MALDI-TOF MS fingerprinting. Electrophoresis 2013, 34, 877–887. [Google Scholar] [CrossRef]
- Adewoyin, M.A.; Okoh, A.I. The natural environment as a reservoir of pathogenic and non-pathogenic Acinetobacter species. Rev. Environ. Health 2018, 33, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Mukherjee, S.; Rana, V.S.; Saha, D.R.; Raman, R.; Padhy, P.K.; Mazumder, S. Isolation and characterization of arsenic-resistant bacteria from contaminated water-bodies in West Bengal, India. Geomicrobiol. J. 2015, 32, 17–26. [Google Scholar] [CrossRef]
- Guardabassi, L.; Dalsgaard, A.; Olsen, J.E. Phenotypic characterization and antibiotic resistance of Acinetobacter spp. isolated from aquatic sources. J. Appl. Microbiol. 1999, 87, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Roseline, T.L.; Sachindra, N.M. Characterization of extracellular agarase production by Acinetobacter junii PS12B, isolated from marine sediments. Biocatal. Agric. Biotechnol. 2016, 6, 219–226. [Google Scholar] [CrossRef]
- Weidmann-Al-Ahmad, M.; Tichy, H.V.; Schön, G. Characterisation of Acinetobacter type strains and isolates obtained from wastewater treatment plants by PCR fingerprinting. Appl. Environ. Microbiol. 1994, 60, 4066–4071. [Google Scholar] [CrossRef]
- Rafei, R.; Hamze, M.; Pailhoriès, H.; Eveillard, M.; Marsollier, L.; Joly-Guillou, M.L.; Dabboussi, F.; Kempf, M. Extrahuman epidemiology of Acinetobacter baumannii in Lebanon. Appl. Environ. Microbiol. 2015, 81, 2359–2367. [Google Scholar] [CrossRef]
- Sunantaraporn, S.; Sanprasert, V.; Pengsakul, T.; Phumee, A.; Boonserm, R.; Tawatsin, A.; Thavara, U.; Siriyasatien, P. Molecular survey of the head louse Pediculushumanus capitis in Thailand and its potential role for transmitting Acinetobacter spp. Parasites Vectors 2015, 8, 127. [Google Scholar] [CrossRef]
- Choi, J.Y.; Kim, Y.; Ko, E.A.; Park, Y.K.; Jheong, W.-H.; Ko, G.P.; Ko, K.S. Acinetobacter species isolates from a range of environments: Species survey and observations of antimicrobial resistance. Diagn. Micr. Infec. Dis. 2012, 74, 177–180. [Google Scholar] [CrossRef]
- Carvalheira, A.; Casquete, R.; Silva, J.; Teixeira, P. Prevalence and antimicrobial susceptibility of Acinetobacter spp. isolated from meat. Int. J. Food Microbiol. 2017, 243, 58–63. [Google Scholar] [CrossRef]
- Dijkshoorn, L.; van Aken, E.; Shunburne, L.; van der Reijden, T.J.; Bernards, A.T.; Nemec, A.; Towner, K.J. Prevalence of Acinetobacter baumannii and other Acinetobacter spp. in faecal samples from non-hospitalised individuals. Clin. Microbiol. Infect. 2005, 11, 329–332. [Google Scholar] [CrossRef]
- Lehosmaa, K.; Muotka, T.; Pirttilä, A.M.; Jaakola, I.; Rossi, P.M.; Jyväsjärvi, J. Bacterial communities at a groundwater-surface water ecotone: Gradual change or abrupt transition points along a contamination gradient? Environ. Microbiol. 2021, 23, 6694–6706. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| 16S rRNA Gene Sequencing Identification | MALDI-TOF MS Identification | ||||||
|---|---|---|---|---|---|---|---|
| Bacterial Genus | Number of Isolates | Species Level > 98.5% | Genus Level > 95% | No Identification < 95% | Species Level > 2 | Genus Level 2 > 1.7 | No Identification <1.7 |
| Acinetobacter | 20 | 16 | 2 | 2 | 16 | 4 | |
| Aeromonas | 1 | 1 | 1 | ||||
| Brevundimonas | 3 | 3 | 3 | ||||
| Chryseobacterium | 1 | 1 | 1 | ||||
| Enterobacter | 5 | 1 | 4 | 2 | 3 | ||
| Microbacterium | 1 | 1 | 1 | ||||
| Pantoea | 1 | 1 | 1 | ||||
| Pseudarthrobacter | 1 | 1 | 1 | ||||
| Pseudomonas | 5 | 2 | 3 | 3 | 1 | 1 | |
| Rhodococcus | 2 | 1 | 1 | 2 | |||
| Sphingobacterium | 1 | 1 | 1 | ||||
| Stenotrophomonas | 1 | 1 | 1 | ||||
| Total isolates | 42 | 27 (64.3%) | 38 (90.5%) | 4 (9.5%) | 28 (66.7%) | 40 (95.2%) | 2 (4.8%) |
| No. | Isolate | MALDI-TOF MS Identification (Log Score, Consistency Category) | 16S rRNA Identification (% Similarity Score) |
|---|---|---|---|
| #1 | Sample5/9 | Acinetobacter junii (2.34; A) | Acinetobacter schindleri (99.24%) |
| #2 | Sample5/12 | Acinetobacter junii (2.1; A) | Acinetobacter schindleri (98.78%) |
| #3 | Sample3/1 | Rhodococcus spp. (1.71; B) | Rhodococcus qinsenghii (96.2%) |
| #4 | Sample3/3 | No ID (1.51; C) | Sphingobacterium kitahiroshimense (99.72%) |
| #5 | Sample3/4 | Chryseobacterium indologenes (2.01; A) | Chryseobacterium lactis (98.8%) |
| #6 | Sample2/4 | Enterobacter hormaechei (2.25; A) | Enterobacter cloacae/E. hormaechei (99.9%) |
| #7 | Sample2/5 | Pseudarthrobacter scleromae/oxydans (2.24; B) | Pseudarthrobacter siccitolerans (89.91%) |
| #8 | Sample2/6 | Rhodococcus spp. (1.99; B) | Rhodococcus cerastii (99.46%) |
| #9 | Sample2/7 | Enterobacter cloacae (2.27; A) | E. hormacheai (99.48%) |
| #10 | Sample2/8 | Pseudomonas veronii (2.26; A) | P. veronii/ P. extremaustralis (100%) |
| #11 | Sample2/9 | Pseudomonas veronii (2.2; A) | Pseudomonas spp. (99.34%) |
| Bacterial Genus | Number of Isolates | Relative Abundance of the Genera |
|---|---|---|
| Brevundimonas | 3 | 2.18% |
| Rhodococcus | 2 | 0.81% |
| Acinetobacter | 20 | 0.64%. |
| Chryseobacterium | 1 | 0.35% |
| Pseudomonas | 5 | 0.24% |
| Enterobacter | 5 | 0.04% |
| Stenotrophomonas | 1 | 0.03% |
| Sphingobacterium | 1 | 0.02% |
| Aeromonas | 1 | <0.01% |
| Microbacterium | 1 | <0.01% |
| Pantoea | 1 | <0.01% |
| Pseudarthrobacter | 1 | <0.01% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surányi, B.B.; Zwirzitz, B.; Mohácsi-Farkas, C.; Engelhardt, T.; Domig, K.J. Comparing the Efficacy of MALDI-TOF MS and Sequencing-Based Identification Techniques (Sanger and NGS) to Monitor the Microbial Community of Irrigation Water. Microorganisms 2023, 11, 287. https://doi.org/10.3390/microorganisms11020287
Surányi BB, Zwirzitz B, Mohácsi-Farkas C, Engelhardt T, Domig KJ. Comparing the Efficacy of MALDI-TOF MS and Sequencing-Based Identification Techniques (Sanger and NGS) to Monitor the Microbial Community of Irrigation Water. Microorganisms. 2023; 11(2):287. https://doi.org/10.3390/microorganisms11020287
Chicago/Turabian StyleSurányi, Botond Bendegúz, Benjamin Zwirzitz, Csilla Mohácsi-Farkas, Tekla Engelhardt, and Konrad J. Domig. 2023. "Comparing the Efficacy of MALDI-TOF MS and Sequencing-Based Identification Techniques (Sanger and NGS) to Monitor the Microbial Community of Irrigation Water" Microorganisms 11, no. 2: 287. https://doi.org/10.3390/microorganisms11020287
APA StyleSurányi, B. B., Zwirzitz, B., Mohácsi-Farkas, C., Engelhardt, T., & Domig, K. J. (2023). Comparing the Efficacy of MALDI-TOF MS and Sequencing-Based Identification Techniques (Sanger and NGS) to Monitor the Microbial Community of Irrigation Water. Microorganisms, 11(2), 287. https://doi.org/10.3390/microorganisms11020287