

Diversity, Taxonomic Novelty, and Encoded Functions of Salar de Ascotán Microbiota, as Revealed by Metagenome-Assembled Genomes
 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Site of Study and Sample Collection
2.2. DNA Purification and Quality Assessment
2.3. 16S rRNA Metabarcoding and Microbial Diversity Analyses
2.4. Illumina and Nanopore Shotgun DNA Sequencing
2.5. Shotgun Sequence Diversity Analyses
2.6. Hybrid Metagenome Assembly and MAGs Reconstruction, Annotation, and Functional Profiling
2.7. Phylogenetic Analyses
3. Results
3.1. Microbial Diversity in Salar de Ascotán’s Water, Sediment, and Soil, as Revealed by 16S rRNA Metabarcoding
3.2. Metagenomic Analyses and Captured Sequence Diversity
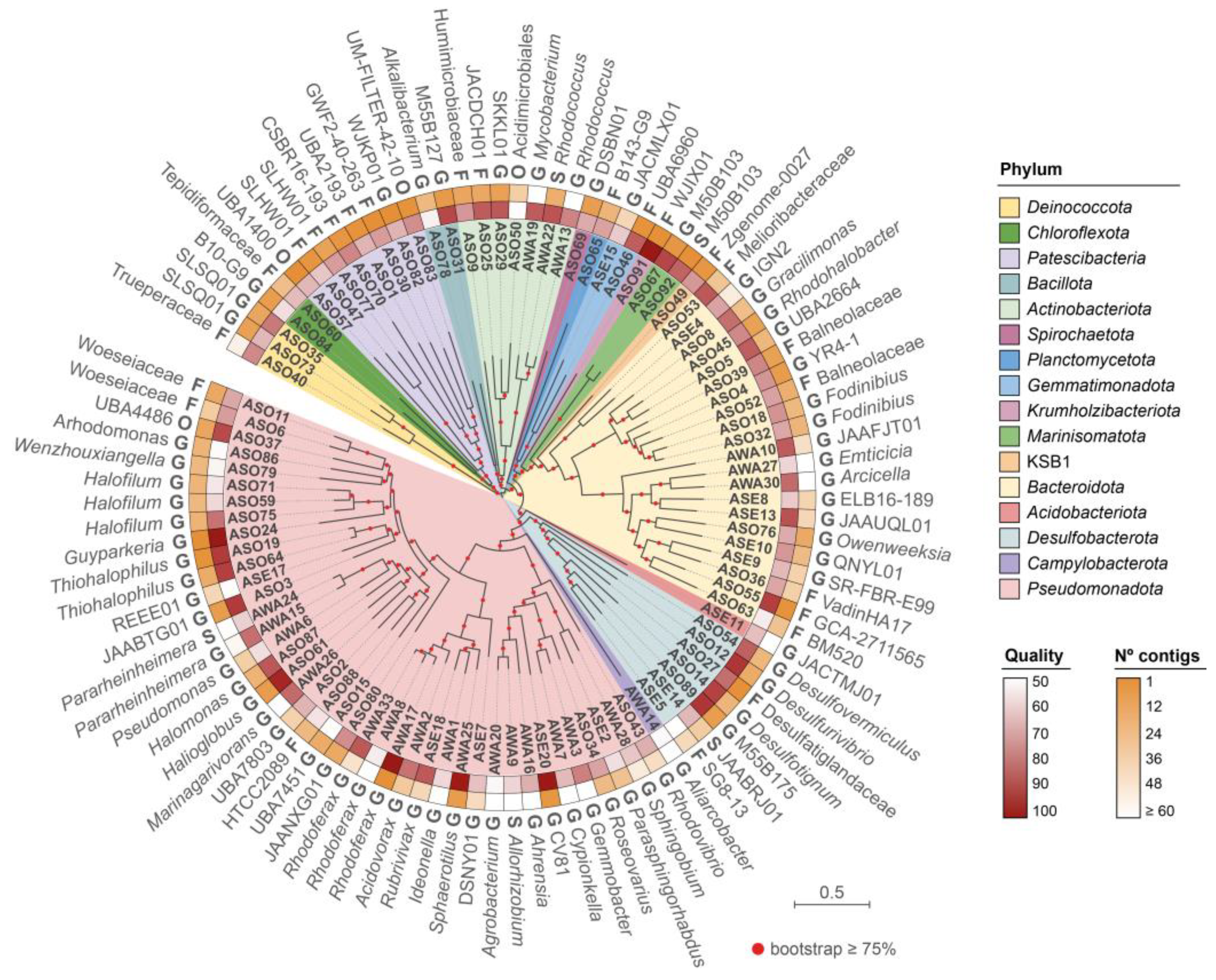
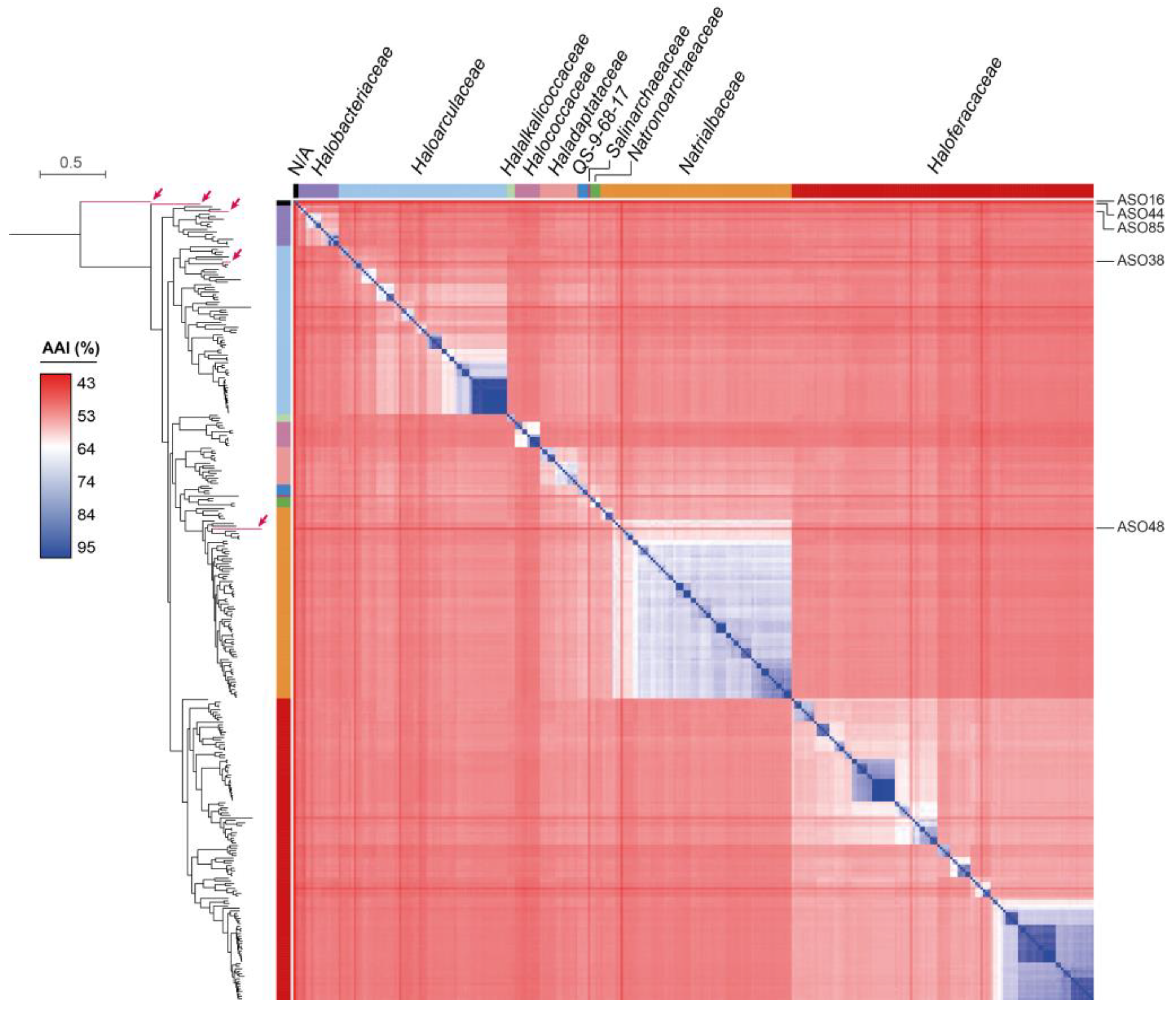
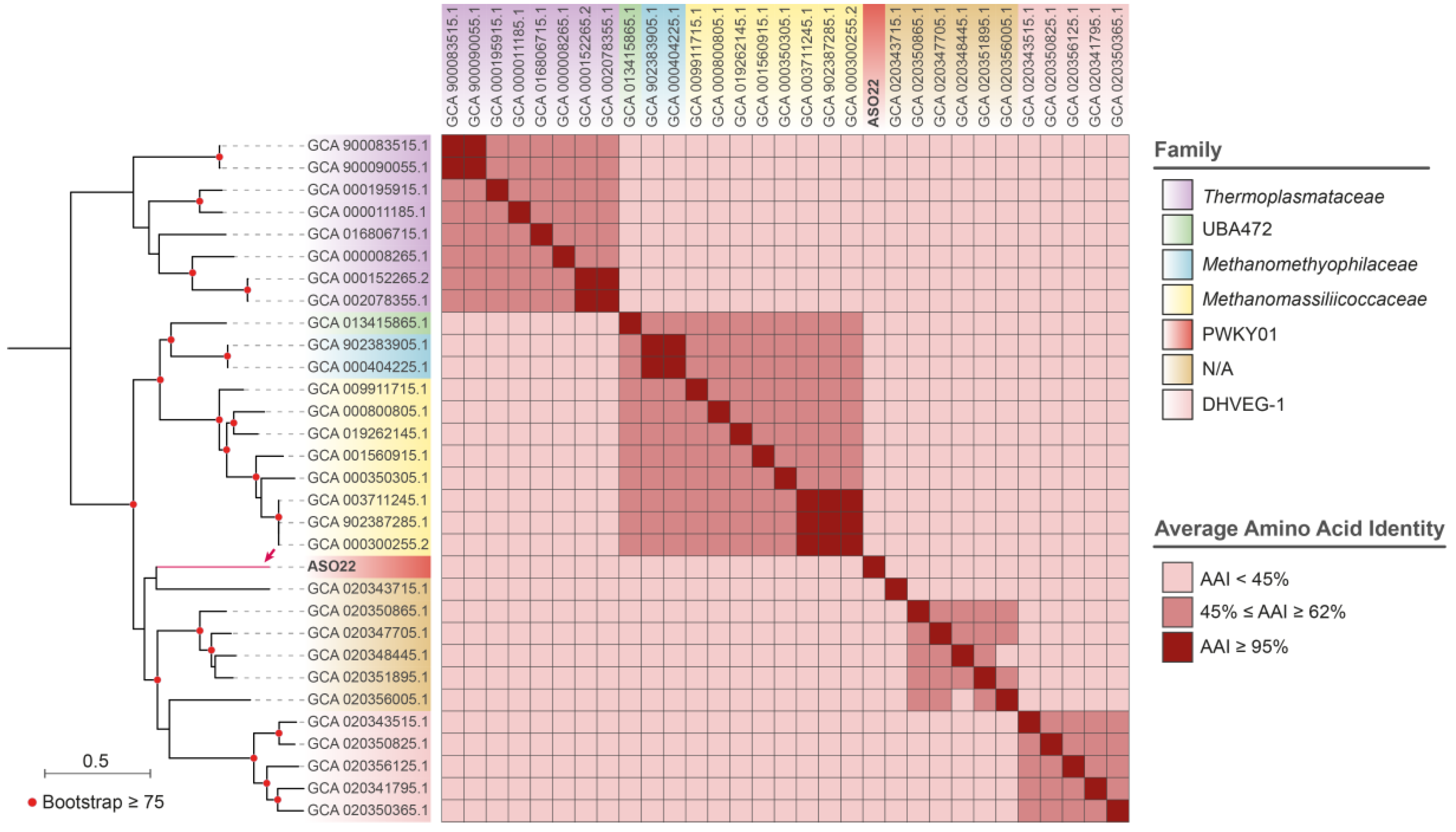
3.3. Reconstruction and Taxonomic Novelty of Bacterial and Archaeal Genomes from Salar de Ascotán
3.4. Predicted Metabolic Capabilities and Molecular Functions Encoded in the Salar de Ascotán’s MAGs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aceituno, P. Elementos Del Clima En El Altiplano Sudamericano. Rev. Geofis. 1996, 44, 37–55. [Google Scholar]
- Cabrol, N.A.; Grin, E.A.; Chong, G.; Minkley, E.; Hock, A.N.; Yu, Y.; Bebout, L.; Fleming, E.; Häder, D.P.; Demergasso, C.; et al. The High-Lakes Project. J. Geophys. Res. Biogeosci. 2009, 114, 1–20. [Google Scholar] [CrossRef]
- Risacher, F.; Alonso, H.; Salazar, C. Geoquímica de Aguas En Cuencas Cerradas: I, II y III Regiones-Chile. Volumen III ESTUDIO DE CUENCAS DE LA II REGION. 1999, pp. 1–299. Available online: https://bibliotecadigital.ciren.cl/handle/20.500.13082/32750 (accessed on 1 September 2023).
- Lara, J.; González, L.E.; Ferrero, M.; Díaz, G.C.; Pedrós-Alió, C.; Demergasso, C. Enrichment of Arsenic Transforming and Resistant Heterotrophic Bacteria from Sediments of Two Salt Lakes in Northern Chile. Extremophiles 2012, 16, 523–538. [Google Scholar] [CrossRef]
- Escudero, L.V.; Casamayor, E.O.; Chong, G.; Pedrós-Alió, C.; Demergasso, C. Distribution of Microbial Arsenic Reduction, Oxidation and Extrusion Genes along a Wide Range of Environmental Arsenic Concentrations. PLoS ONE 2013, 8, e78890. [Google Scholar] [CrossRef] [PubMed]
- Dorador, C. Microbial Communities in High Altitude Altiplanic Wetlands in Northern Chile: Phylogeny, Diversity and Function. Ph.D. Thesis, Christian-Albrechts-Universität, Kiel, Germany, 2007. [Google Scholar]
- Demergasso, C.; Casamayor, E.O.; Chong, G.; Galleguillos, P.; Escudero, L.; Pedrós-Alió, C. Distribution of Prokaryotic Genetic Diversity in Athalassohaline Lakes of the Atacama Desert, Northern Chile. FEMS Microbiol. Ecol. 2004, 48, 57–69. [Google Scholar] [CrossRef]
- Teiller, S.; Becerra, P. Flora and Vegetation of Ascotan Saltmarsh, Northern Chilean Andes. Gayana Bot. 2003, 60, 114–122. [Google Scholar]
- Vila, I.; Morales, P.; Scott, S.; Poulin, E.; Véliz, D.; Harrod, C.; Méndez, M.A. Phylogenetic and Phylogeographic Analysis of the Genus Orestias (Teleostei: Cyprinodontidae) in the Southern Chilean Altiplano: The Relevance of Ancient and Recent Divergence Processes in Speciation. J. Fish Biol. 2013, 82, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Di Genova, A.; Nardocci, G.; Maldonado-Agurto, R.; Hodar, C.; Valdivieso, C.; Morales, P.; Gajardo, F.; Marina, R.; Gutiérrez, R.A.; Orellana, A.; et al. Genome Sequencing and Transcriptomic Analysis of the Andean Killifish Orestias ascotanensis Reveals Adaptation to High-Altitude Aquatic Life. Genomics 2022, 114, 305–315. [Google Scholar] [CrossRef]
- Taş, N.; de Jong, A.E.; Li, Y.; Trubl, G.; Xue, Y.; Dove, N.C. Metagenomic Tools in Microbial Ecology Research. Curr. Opin. Biotechnol. 2021, 67, 184–191. [Google Scholar] [CrossRef]
- Cowan, D.A.; Ramond, J.B.; Makhalanyane, T.P.; De Maayer, P. Metagenomics of Extreme Environments. Curr. Opin. Microbiol. 2015, 25, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Rappé, M.S.; Giovannoni, S.J. The Uncultured Microbial Majority. Annu. Rev. Microbiol. 2003, 57, 369–394. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.L.Z.; Sicheritz-Pontén, T.; Thomas Gilbert, M.P. Environmental Genes and Genomes: Understanding the Differences and Challenges in the Approaches and Software for Their Analyses. Brief. Bioinform. 2014, 16, 745–758. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, L.; Liu, X.; Guan, F.; Xu, Y.; Yue, H.; Huang, J.Q.; Chen, J.; Wu, N.; Tian, J. Combined Assembly of Long and Short Sequencing Reads Improve the Efficiency of Exploring the Soil Metagenome. BMC Genom. 2022, 23, 37. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhao, N.; Cao, J.; Liu, X.; Xu, J.; Ma, Y.; Yu, Y.; Zhang, X.; Zhang, W.; Guan, X.; et al. Short- and Long-Read Metagenomics Expand Individualized Structural Variations in Gut Microbiomes. Nat. Commun. 2022, 13, 3175. [Google Scholar] [CrossRef] [PubMed]
- Zenoff, V.F.; Heredia, J.; Ferrero, M.; Siñeriz, F.; Farías, M.E. Diverse UV-B Resistance of Culturable Bacterial Community from High-Altitude Wetland Water. Curr. Microbiol. 2006, 52, 359–362. [Google Scholar] [CrossRef]
- Demergasso, C.; Dorador, C.; Meneses, D.; Blamey, J.; Cabrol, N.; Escudero, L.; Chong, G. Prokaryotic Diversity Pattern in High-Altitude Ecosystems of the Chilean Altiplano. J. Geophys. Res. Biogeosci. 2010, 115, 1–14. [Google Scholar] [CrossRef]
- Valenzuela, C.; Campos, V.L.; Yañez, J.; Zaror, C.A.; Mondaca, M.A. Isolation of Arsenite-Oxidizing Bacteria from Arsenic-Enriched Sediments from Camarones River, Northern Chile. Bull. Environ. Contam. Toxicol. 2009, 82, 593–596. [Google Scholar] [CrossRef]
- Chou, C.L.; Paon, L.A.; Moffatt, J.D.; Zwicker, B. Copper Contamination and Cadmium, Silver, and Zinc Concentrations in the Digestive Glands of American Lobster (Homarus americanus) from the Inner Bay of Fundy, Atlantic Canada. Bull. Environ. Contam. Toxicol. 2000, 65, 470–477. [Google Scholar] [CrossRef]
- Estaki, M.; Jiang, L.; Bokulich, N.A.; McDonald, D.; González, A.; Kosciolek, T.; Martino, C.; Zhu, Q.; Birmingham, A.; Vázquez-Baeza, Y.; et al. QIIME 2 Enables Comprehensive End-to-End Analysis of Diverse Microbiome Data and Comparative Studies with Publicly Available Data. Curr. Protoc. Bioinform. 2020, 70, 1–46. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and Processing Long-Read Sequencing Data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast Genome and Metagenome Distance Estimation Using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Katz, L.; Griswold, T.; Morrison, S.; Caravas, J.; Zhang, S.; Bakker, H.; Deng, X.; Carleton, H. Mashtree: A Rapid Comparison of Whole Genome Sequence Files. J. Open Source Softw. 2019, 4, 1762. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Gunturu, S.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. Nonpareil 3: Fast Estimation of Metagenomic Coverage and Sequence Diversity. mSystems 2018, 3, e00039-18. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and Sensitive Taxonomic Classification for Metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.L.; et al. MetaFlye: Scalable Long-Read Metagenome Assembly Using Repeat Graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Wick, R.R.; Holt, K.E. Polypolish: Short-Read Polishing of Long-Read Bacterial Genome Assemblies. PLoS Comput. Biol. 2022, 18, e1009802. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA Genome Assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A Flexible Pipeline for Genome-Resolved Metagenomic Data Analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- Alneberg, J.; Bjarnason, B.S.; De Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning Metagenomic Contigs by Coverage and Composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An Automated Binning Algorithm to Recover Genomes from Multiple Metagenomic Datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory Friendly Classification with the Genome Taxonomy Database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, M.; Borton, M.A.; McGivern, B.B.; Zayed, A.A.; La Rosa, S.L.; Solden, L.M.; Liu, P.; Narrowe, A.B.; Rodríguez-Ramos, J.; Bolduc, B.; et al. DRAM for Distilling Microbial Metabolism to Automate the Curation of Microbiome Function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.G.Z.; Green, K.T.; Dutilh, B.E.; Edwards, R.A. SUPER-FOCUS: A Tool for Agile Functional Analysis of Shotgun Metagenomic Data. Bioinformatics 2016, 32, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.G.J. BacMet: Antibacterial Biocide and Metal Resistance Genes Database. Nucleic Acids Res. 2014, 42, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Gunturu, S.; Harvey, W.T.; Rosselló-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) Webserver: Taxonomic and Gene Diversity Analysis of Archaea and Bacteria at the Whole Genome Level. Nucleic Acids Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef]
- Yu, Y.; Lee, C.; Kim, J.; Hwang, S. Group-Specific Primer and Probe Sets to Detect Methanogenic Communities Using Quantitative Real-Time Polymerase Chain Reaction. Biotechnol. Bioeng. 2005, 89, 670–679. [Google Scholar] [CrossRef]
- Nehmé, B.; Gilbert, Y.; Létourneau, V.; Forster, R.J.; Veillette, M.; Villemur, R.; Duchaine, C. Culture-Independent Characterization of Archaeal Biodiversity in Swine Confinement Building Bioaerosols. Appl. Environ. Microbiol. 2009, 75, 5445–5450. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.H.; Farag, I.F.; Hahn, C.R.; Premathilake, H.; Fry, E.; Hart, M.; Huffaker, K.; Bird, E.; Hambright, J.; Hoff, W.D.; et al. Candidatus Krumholzibacterium Zodletonense gen. nov., sp nov, the First Representative of the Candidate Phylum Krumholzibacteriota Phyl. Nov. Recovered from an Anoxic Sulfidic Spring Using Genome Resolved Metagenomics. Syst. Appl. Microbiol. 2019, 42, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Tarn, J.; Peoples, L.M.; Hardy, K.; Cameron, J.; Bartlett, D.H. Identification of Free-Living and Particle-Associated Microbial Communities Present in Hadal Regions of the Mariana Trench. Front. Microbiol. 2016, 7, 665. [Google Scholar] [CrossRef] [PubMed]
- Pérez Castro, S.; Borton, M.A.; Regan, K.; Hrabe de Angelis, I.; Wrighton, K.C.; Teske, A.P.; Strous, M.; Ruff, S.E. Degradation of Biological Macromolecules Supports Uncultured Microbial Populations in Guaymas Basin Hydrothermal Sediments. ISME J. 2021, 15, 3480–3497. [Google Scholar] [CrossRef] [PubMed]
- Suarez, C.; Hackl, T.; Wilen, B.-M.; Persson, F.; Hagelia, P.; Jetten, M.; Martins, P.D. Novel and Unusual Genes for Nitrogen and Metal Cycling in Planctomycetota- and KSB1-Affiliated Metagenome-Assembled Genomes Reconstructed from a Marine Subsea Tunnel. FEMS Microbiol. Lett. 2023, 370, fnad049. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, Y.; Lu, R.; Zheng, P.; Wang, Y. Phylogeny, Distribution and Potential Metabolism of Candidate Bacterial Phylum KSB1. PeerJ 2022, 10, e13241. [Google Scholar] [CrossRef]
- Ji, Y.; Zhang, P.; Zhou, S.; Gao, P.; Wang, B.; Jiang, J. Widespread but Poorly Understood Bacteria: Candidate Phyla Radiation. Microorganisms 2022, 10, 2232. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T.; Hug, L.A.; Thomas, B.C.; Sharon, I.; Castelle, C.J.; Singh, A.; Wilkins, M.J.; Wrighton, K.C.; Williams, K.H.; Banfield, J.F. Unusual Biology across a Group Comprising More than 15% of Domain Bacteria. Nature 2015, 523, 208–211. [Google Scholar] [CrossRef]
- Jackson, K.L.; Whitcraft, C.R.; Dillon, J.G. Diversity of Desulfobacteriaceae and Overall Activity of Sulfate-Reducing Microorganisms in and around a Salt Pan in a Southern California Coastal Wetland. Wetlands 2014, 34, 969–977. [Google Scholar] [CrossRef]
- Caffrey, J.M.; Bano, N.; Kalanetra, K.; Hollibaugh, J.T. Ammonia Oxidation and Ammonia-Oxidizing Bacteria and Archaea from Estuaries with Differing Histories of Hypoxia. ISME J. 2007, 1, 660–662. [Google Scholar] [CrossRef] [PubMed]
- Li, L.G.; Xia, Y.; Zhang, T. Co-Occurrence of Antibiotic and Metal Resistance Genes Revealed in Complete Genome Collection. ISME J. 2017, 11, 651–662. [Google Scholar] [CrossRef]
- Kurth, D.; Amadio, A.; Ordoñez, O.F.; Albarracín, V.H.; Gärtner, W.; Farías, M.E. Arsenic Metabolism in High Altitude Modern Stromatolites Revealed by Metagenomic Analysis. Sci. Rep. 2017, 7, 1024. [Google Scholar] [CrossRef]
- Hobman, J.L.; Crossman, L.C. Bacterial Antimicrobial Metal Ion Resistance. J. Med. Microbiol. 2015, 64, 471–497. [Google Scholar] [CrossRef] [PubMed]
- Altermann, E. Tracing Lifestyle Adaptation in Prokaryotic Genomes. Front. Microbiol. 2012, 3, 48. [Google Scholar] [CrossRef]
- Dorador, C.; Vila, I.; Witzel, K.P.; Imhof, J.F. Bacterial and Archaeal Diversity in High Altitude Wetlands of the Chilean Altiplano. Fundam. Appl. Limnol. 2013, 182, 135–159. [Google Scholar] [CrossRef]
- Demergasso, C.; Escudero, L.; Casamayor, E.O.; Chong, G.; Balagué, V.; Pedrós-Alió, C. Novelty and Spatio-Temporal Heterogeneity in the Bacterial Diversity of Hypersaline Lake Tebenquiche (Salar de Atacama). Extremophiles 2008, 12, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Dorador, C.; Vila, I.; Remonsellez, F.; Imhoff, J.F.; Witzel, K.P. Unique Clusters of Archaea in Salar de Huasco, an Athalassohaline Evaporitic Basin of the Chilean Altiplano. FEMS Microbiol. Ecol. 2010, 73, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Gong, J. A Meta-Analysis of the Publicly Available Bacterial and Archaeal Sequence Diversity in Saline Soils. World J. Microbiol. Biotechnol. 2013, 29, 2325–2334. [Google Scholar] [CrossRef]
- Martin, E.L.; Reinhardt, R.L.; Baum, L.L.; Becker, M.R.; Shaffer, J.J.; Kokjohn, T.A. The Effects of Ultraviolet Radiation on the Moderate Halophile Halomonas elongata and the Extreme Halophile Halobacterium salinarum. Can. J. Microbiol. 2000, 46, 180–187. [Google Scholar] [CrossRef]
- Kim, K.K.; Lee, J.S.; Stevens, D.A. Microbiology and Epidemiology of Halomonas Species. Future Microbiol. 2013, 8, 1559–1573. [Google Scholar] [CrossRef] [PubMed]
- Bird, L.J.; Mickol, R.L.; Eddie, B.J.; Thakur, M.; Yates, M.D.; Glaven, S.M. Marinobacter: A Case Study in Bioelectrochemical Chassis Evaluation. Microb. Biotechnol. 2023, 16, 494–506. [Google Scholar] [CrossRef]
- He, Y.; He, L.; Wang, Z.; Liang, T.; Sun, S.; Liu, X. Salinity Shapes the Microbial Communities in Surface Sediments of Salt Lakes on the Tibetan Plateau, China. Water 2022, 14, 4043. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Kuenen, J.G. Haloalkaliphilic Sulfur-Oxidizing Bacteria in Soda Lakes. FEMS Microbiol. Rev. 2005, 29, 685–702. [Google Scholar] [CrossRef]
- Bowman, J.P.; McCammon, S.A.; Lewis, T.; Skerratt, J.H.; Brown, J.L.; Nichols, D.S.; McMeekin, T.A. Psychroflexus torquis gen. nov., sp. nov. a Psychrophilic Species from Antarctic Sea Ice, and Reclassification of Flavobacterium Gondwanense (Dobson et Al. 1993) as Psychroflexus gondwanense gen. nov., comb. nov. Microbiology 1998, 144, 1601–1609. [Google Scholar] [CrossRef]
- Dorador, C.; Meneses, D.; Urtuvia, V.; Demergasso, C.; Vila, I.; Witzel, K.P.; Imhoff, J.F. Diversity of Bacteroidetes in High-Altitude Saline Evaporitic Basins in Northern Chile. J. Geophys. Res. Biogeosci. 2009, 114, 1–11. [Google Scholar] [CrossRef]
- Cho, G.Y.; Lee, J.C.; Whang, K.S. Aliifodinibius salicampi sp. nov., a Moderately Halophilic Bacterium Isolated from a Grey Saltern. Int. J. Syst. Evol. Microbiol. 2017, 67, 2598–2603. [Google Scholar] [CrossRef]
- Cho, G.Y.; Whang, K.S. Aliifodinibius saliphilus sp. Nov., a Moderately Halophilic Bacterium Isolated from Sediment of a Crystallizing Pond of a Saltern. Int. J. Syst. Evol. Microbiol. 2020, 70, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Liu, J.H.; Xiao, W.; Ma, X.L.; Lai, Y.H.; Li, Z.Y.; Ji, K.Y.; Wen, M.L.; Cui, X.L. Aliifodinibius roseus gen. nov., sp. nov., and Aliifodinibius sediminis sp. nov., Two Moderately Halophilic Bacteria Isolated from Salt Mine Samples. Int. J. Syst. Evol. Microbiol. 2013, 63, 2907–2913. [Google Scholar] [CrossRef] [PubMed]
- Abdeljabbar, H.; Cayol, J.-L.; Hania, W.B.; Boudabous, A.; Sadfi, N.; Fardeau, M.-L. Halanaerobium sehlinense sp. nov., an Extremely Halophilic, Fermentative, Strictly Anaerobic Bacterium from Sediments of the Hypersaline Lake Sehline Sebkha. Int. J. Syst. Evol. Microbiol. 2013, 63, 2069–2074. [Google Scholar] [CrossRef]
- Boltyanskaya, Y.; Zhilina, T.; Grouzdev, D.; Detkova, E.; Pimenov, N.; Kevbrin, V. Halanaerobium polyolivorans sp. nov.—A Novel Halophilic Alkalitolerant Bacterium Capable of Polyol Degradation: Physiological Properties and Genomic Insights. Microorganisms 2023, 11, 2325. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. Euryarchaeota. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2019; pp. 1–17. ISBN 9780470015902. [Google Scholar]
- Oren, A. The Family Halobacteriaceae. In The Prokaryotes: Other Major Lineages of Bacteria and the Archaea; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 41–121. ISBN 978-3-642-38954-2. [Google Scholar]
- Liu, G.; Gong, Z.; Li, Q. Variations in Gut Bacterial Communities between Lesser White-Fronted Geese Wintering at Caizi and Shengjin Lakes in China. Microbiologyopen 2020, 9, e1037. [Google Scholar] [CrossRef]
- Zhang, S. Altered Gut Archaeal Communities in Anser Erythropus Populations Wintering at Shengjin and Caizi Lakes in China. Pak. J. Zool. 2023, 1–10. [Google Scholar] [CrossRef]
- Eissler, Y.; Dorador, C.; Kieft, B.; Molina, V.; Hengst, M. Virus and Potential Host Microbes from Viral-Enriched Metagenomic Characterization in the High-Altitude Wetland, Salar de Huasco, Chile. Microorganisms 2020, 8, 1077. [Google Scholar] [CrossRef]
- Contreras, F.; Amenabar, M.J.; Blamey, J.M. Purification and Characterization of Two Thermostable Xylanases from a Halotolerant Bacillus sp. Asc6BA Isolated from Salar de Ascotán, Atacama Desert. Extremophiles 2021, 25, 51–59. [Google Scholar] [CrossRef]
- Blum, J.S.; Hernandez-Maldonado, J.; Redford, K.; Sing, C.; Bennett, S.C.; Saltikov, C.W.; Oremland, R.S. Arsenate-Dependent Growth Is Independent of an Arra Mechanism of Arsenate Respiration in the Termite Hindgut Isolate Citrobacter sp. Strain TSA-1. Can. J. Microbiol. 2018, 64, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Castro-Severyn, J.; Remonsellez, F.; Valenzuela, S.L.; Salinas, C.; Fortt, J.; Aguilar, P.; Pardo-Esté, C.; Dorador, C.; Quatrini, R.; Molina, F.; et al. Comparative Genomics Analysis of a New Exiguobacterium Strain from Salar de Huasco Reveals a Repertoire of Stress-Related Genes and Arsenic Resistance. Front. Microbiol. 2017, 8, 456. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Water | Sediment | Soil |
|---|---|---|---|
| Location | S 21°29′52.7″ WO 68°15′25.3″ | S 21°29′52.3″ WO 68°15′26.6″ | S 21°29′53.7″ WO 68°15′26.9″’ |
| Elevation (m.a.s.l 1) | 3343 | 3350 | 3329 |
| Environmental temperature (°C) | 23.1 | 20.5 | 20.5 |
| Water temperature (°C) | 16 | 23.5 | - |
| Pressure (HPa) | 651.9 | 651.3 | 651.3 |
| Humidity (%) | 9.2 | 12.0 | 12.0 |
| Soil or sediment temperature (°C) | - | 22.0 | 17.5 |
| pH | 6.5 | 7.5 | 7.0 |
| Water conductivity (mS) | 3.93 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veloso, M.; Waldisperg, A.; Arros, P.; Berríos-Pastén, C.; Acosta, J.; Colque, H.; Varas, M.A.; Allende, M.L.; Orellana, L.H.; Marcoleta, A.E. Diversity, Taxonomic Novelty, and Encoded Functions of Salar de Ascotán Microbiota, as Revealed by Metagenome-Assembled Genomes. Microorganisms 2023, 11, 2819. https://doi.org/10.3390/microorganisms11112819
Veloso M, Waldisperg A, Arros P, Berríos-Pastén C, Acosta J, Colque H, Varas MA, Allende ML, Orellana LH, Marcoleta AE. Diversity, Taxonomic Novelty, and Encoded Functions of Salar de Ascotán Microbiota, as Revealed by Metagenome-Assembled Genomes. Microorganisms. 2023; 11(11):2819. https://doi.org/10.3390/microorganisms11112819
Chicago/Turabian StyleVeloso, Marcelo, Angie Waldisperg, Patricio Arros, Camilo Berríos-Pastén, Joaquín Acosta, Hazajem Colque, Macarena A. Varas, Miguel L. Allende, Luis H. Orellana, and Andrés E. Marcoleta. 2023. "Diversity, Taxonomic Novelty, and Encoded Functions of Salar de Ascotán Microbiota, as Revealed by Metagenome-Assembled Genomes" Microorganisms 11, no. 11: 2819. https://doi.org/10.3390/microorganisms11112819
APA StyleVeloso, M., Waldisperg, A., Arros, P., Berríos-Pastén, C., Acosta, J., Colque, H., Varas, M. A., Allende, M. L., Orellana, L. H., & Marcoleta, A. E. (2023). Diversity, Taxonomic Novelty, and Encoded Functions of Salar de Ascotán Microbiota, as Revealed by Metagenome-Assembled Genomes. Microorganisms, 11(11), 2819. https://doi.org/10.3390/microorganisms11112819