
Enrichment Pretreatment Expands the Microbial Diversity Cultivated from Marine Sediments
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Bacterial Cultivation
2.2.1. Direct Cultivation without Enrichment Pretreatment
2.2.2. Cultivation with Enrichment Pretreatment
2.3. Bacterial Isolation and Identification
2.4. Amplicon Sequencing and Analyses
2.5. Classification Based on the 16S rRNA Gene
2.6. Testing of Cellulose and Chitin-Degradation Ability
2.7. Genome Sequencing and Analyses
2.8. Statistical Analyses
2.9. Data Availability
3. Results
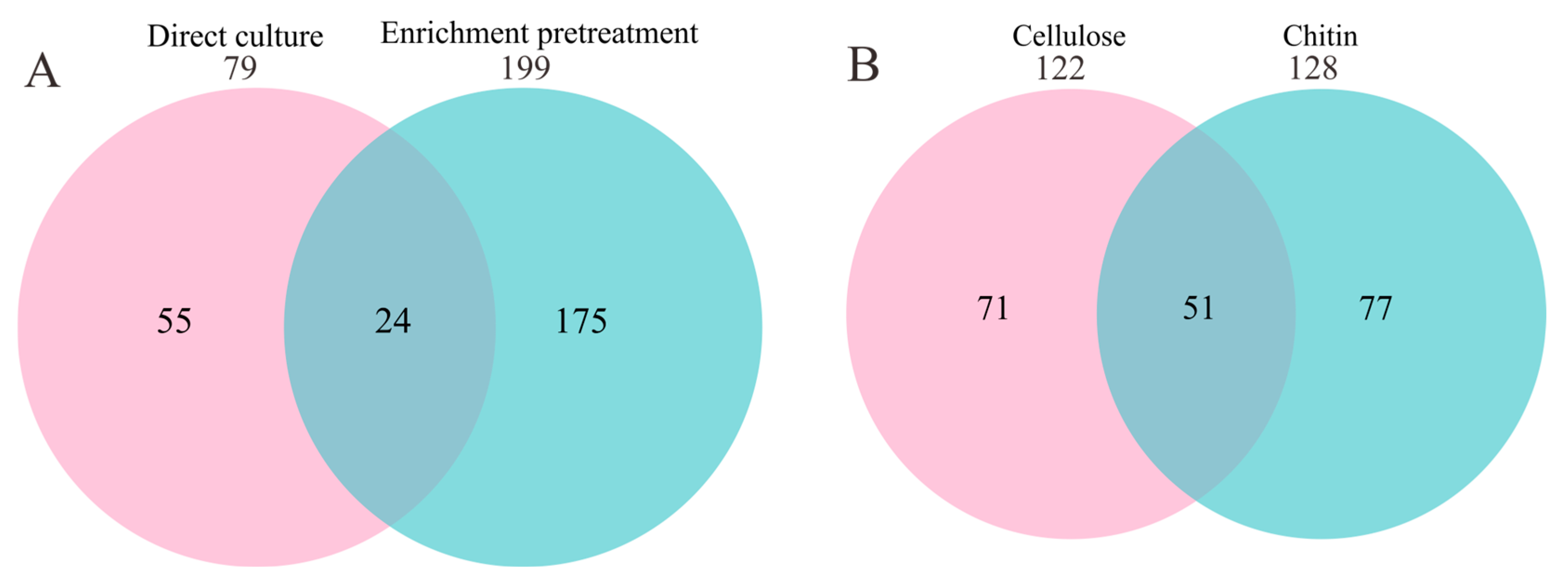
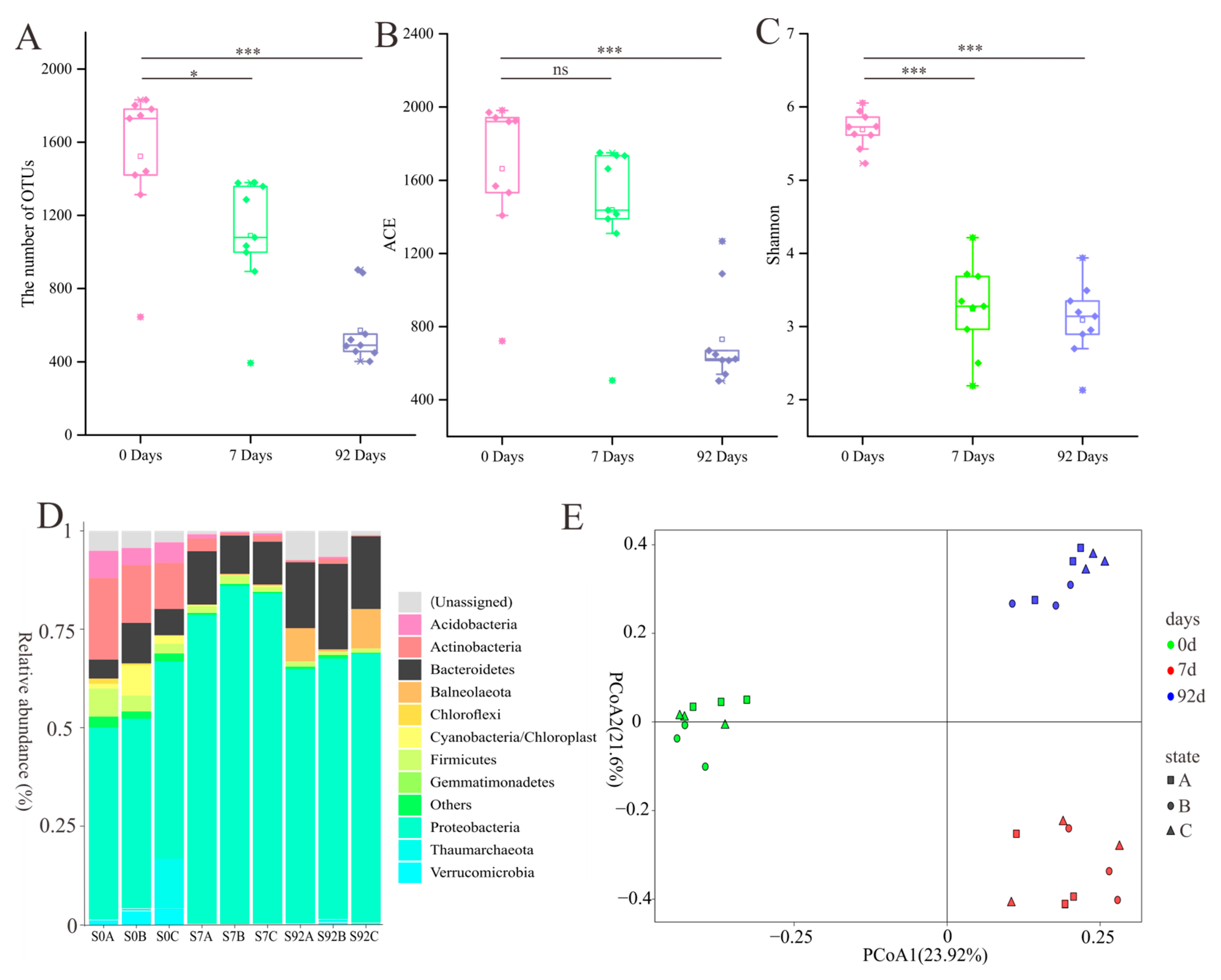
3.1. Large Difference in Diversity Cultivated with and without Enrichment Pretreatment
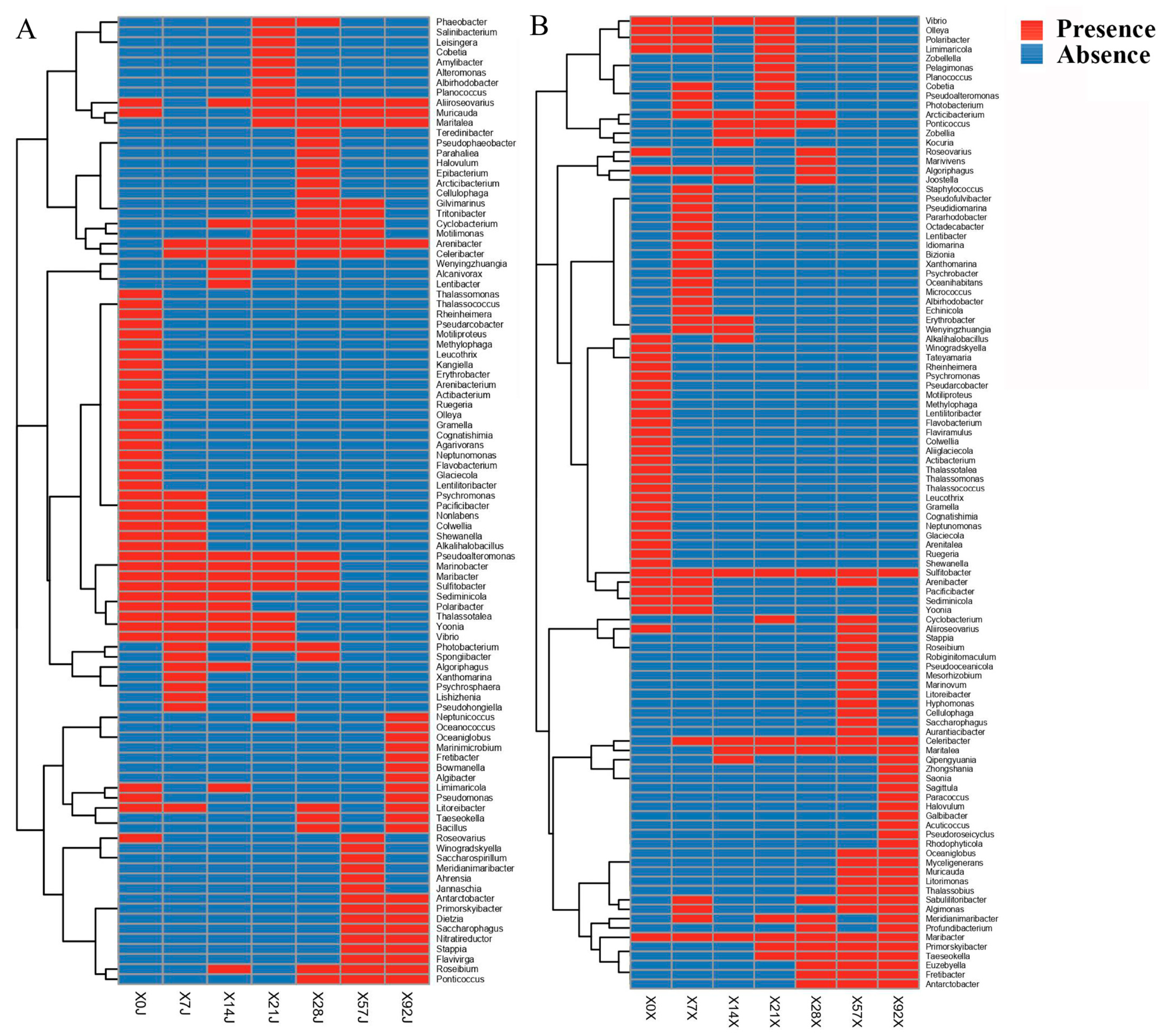
3.2. Different Novel Species Recovered at Different Time Points of Enrichment
3.3. Dynamics of Bacterial Community in the Chitin Enrichment Medium
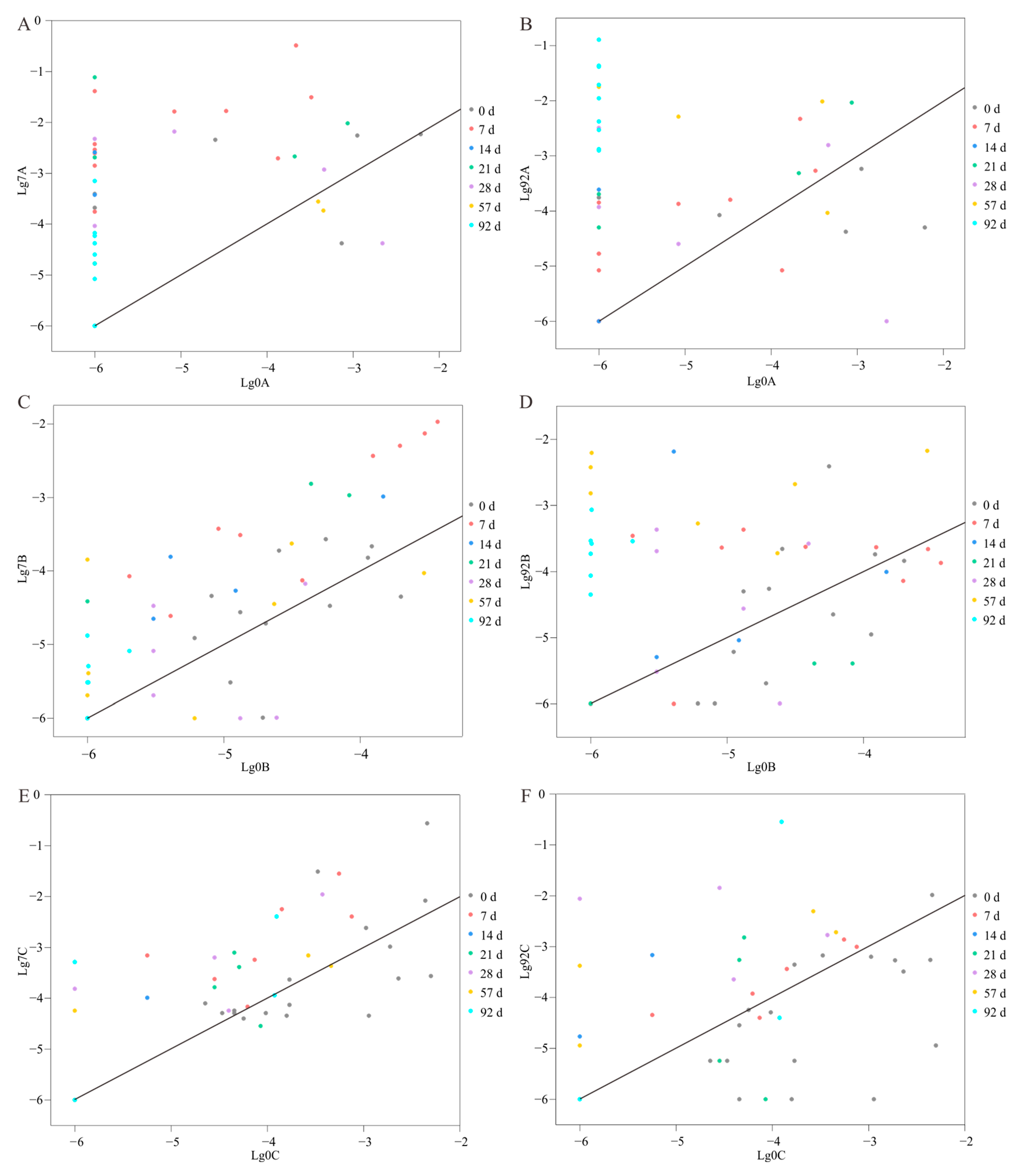
3.4. Increased Relative Abundance of Cultivated Species during Enrichment
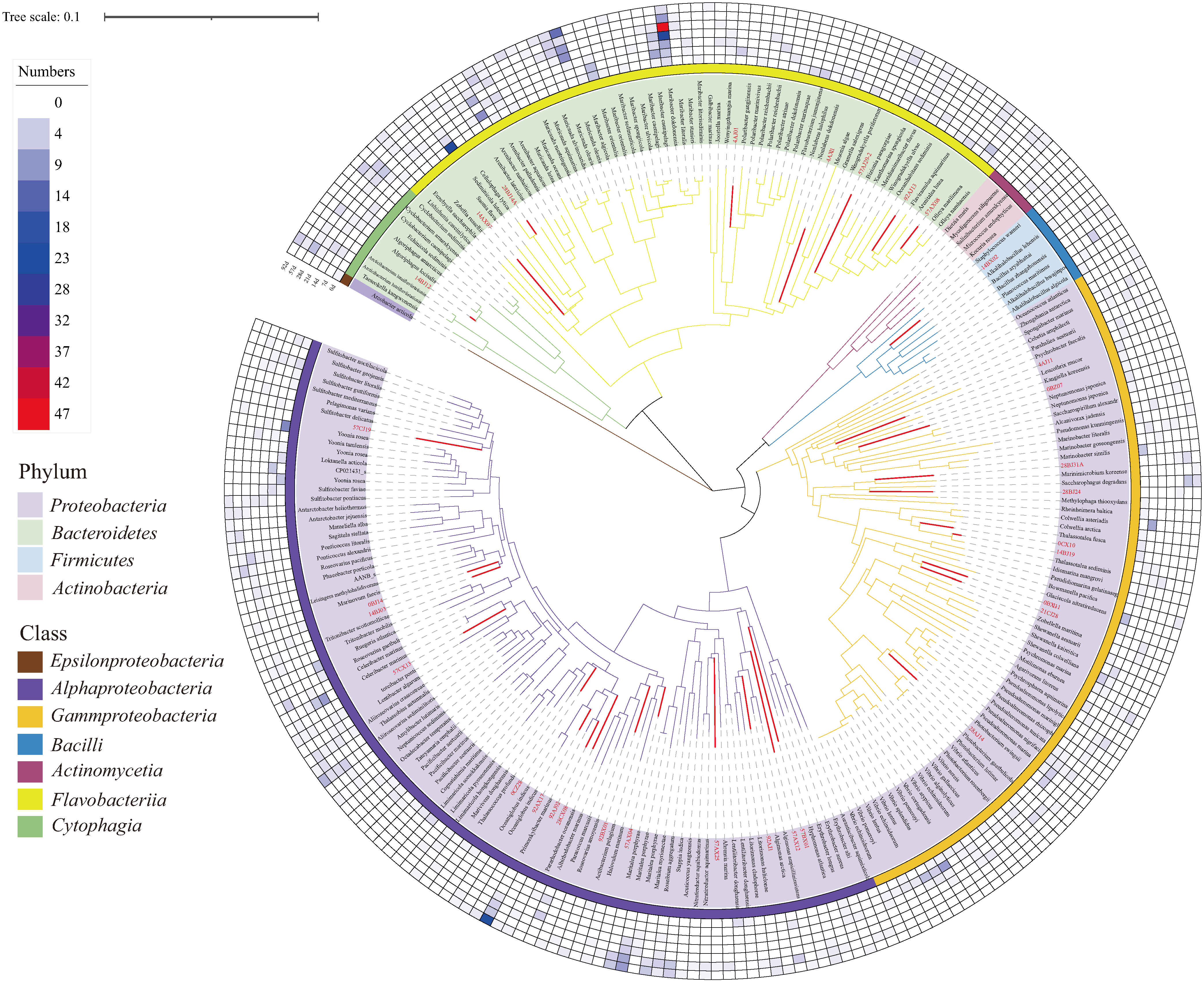
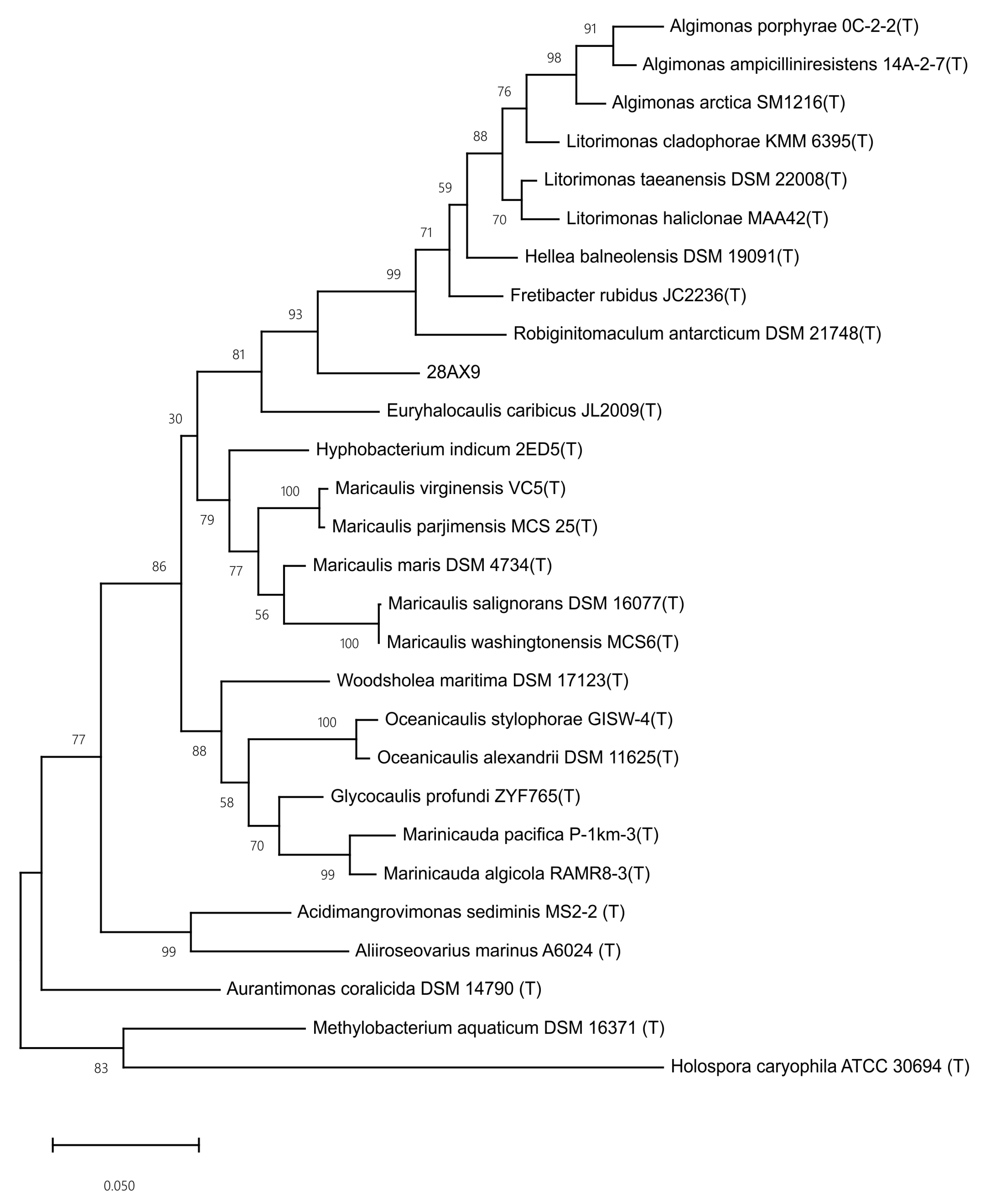
3.5. Genome Sequencing and Phylogenomic Analyses of Novel Species
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Steen, A.D.; Crits-Christoph, A.; Carini, P.; DeAngelis, K.M.; Fierer, N.; Lloyd, K.G.; Cameron Thrash, J. High Proportions of Bacteria and Archaea across Most Biomes Remain Uncultured. ISME J. 2019, 13, 3126–3130. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, K.G.; Steen, A.D.; Ladau, J.; Yin, J.; Crosby, L. Phylogenetically Novel Uncultured Microbial Cells Dominate Earth Microbiomes. mSystems 2018, 3, e00055-18. [Google Scholar] [CrossRef]
- Lewis, W.H.; Tahon, G.; Geesink, P.; Sousa, D.Z.; Ettema, T.J.G. Innovations to Culturing the Uncultured Microbial Majority. Nat. Rev. Microbiol. 2021, 19, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, M.; Huang, L.; Zhang, X.-H. Cultivation of Uncultured Marine Microorganisms. Mar. Life Sci. Technol. 2021, 3, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kawasaki, K.; Daimon, S.; Kitagawa, W.; Yamamoto, K.; Tamaki, H.; Tanaka, M.; Nakatsu, C.H.; Kamagata, Y. A Hidden Pitfall in the Preparation of Agar Media Undermines Microorganism Cultivability. Appl. Environ. Microbiol. 2014, 80, 7659–7666. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Yamagishi, A.; Daimon, S.; Kawasaki, K.; Tamaki, H.; Kitagawa, W.; Abe, A.; Tanaka, M.; Sone, T.; Asano, K.; et al. Isolation of Previously Uncultured Slow-Growing Bacteria by Using a Simple Modification in the Preparation of Agar Media. Appl. Environ. Microbiol. 2018, 84, e00807-18. [Google Scholar] [CrossRef]
- Kaeberlein, T.; Lewis, K.; Epstein, S.S. Isolating “Uncultivable” Microorganisms in Pure Culture in a Simulated Natural Environment. Science 2002, 296, 1127–1129. [Google Scholar] [CrossRef]
- Hu, B.; Xu, P.; Ma, L.; Chen, D.; Wang, J.; Dai, X.; Huang, L.; Du, W. One Cell at a Time: Droplet-Based Microbial Cultivation, Screening and Sequencing. Mar. Life Sci. Technol. 2021, 3, 169–188. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Seck, E.H.; et al. Culture of Previously Uncultured Members of the Human Gut Microbiota by Culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Dubourg, G.; Million, M.; Cadoret, F.; Bilen, M.; Fenollar, F.; Levasseur, A.; Rolain, J.-M.; Fournier, P.-E.; Raoult, D. Culturing the Human Microbiota and Culturomics. Nat. Rev. Microbiol. 2018, 16, 540–550. [Google Scholar] [CrossRef]
- Cross, K.L.; Campbell, J.H.; Balachandran, M.; Campbell, A.G.; Cooper, C.J.; Griffen, A.; Heaton, M.; Joshi, S.; Klingeman, D.; Leys, E.; et al. Targeted Isolation and Cultivation of Uncultivated Bacteria by Reverse Genomics. Nat. Biotechnol. 2019, 37, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lian, Z.; Liu, L.; Fang, B.; Li, W.; Jiao, J. Cultivation Strategies for Prokaryotes from Extreme Environments. iMeta 2023, 2, e123. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Lian, Z.-H.; Narsing Rao, M.P.; Wang, P.; Liu, L.; Fang, B.-Z.; Li, M.-M.; Liu, Z.-T.; Lv, A.-P.; Tan, S.; et al. Insights into the Effects of Drying Treatments on Cultivable Microbial Diversity of Marine Sediments. Microbiol. Res. 2023, 266, 127214. [Google Scholar] [CrossRef]
- Dubourg, G.; Lagier, J.C.; Robert, C.; Armougom, F.; Hugon, P.; Metidji, S.; Dione, N.; Dangui, N.P.M.; Pfleiderer, A.; Abrahao, J.; et al. Culturomics and Pyrosequencing Evidence of the Reduction in Gut Microbiota Diversity in Patients with Broad-Spectrum Antibiotics. Int. J. Antimicrob. Agents 2014, 44, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Oliveira, T.; Wollweber, F.; Ponce-Toledo, R.I.; Xu, J.; Rittmann, S.K.-M.R.; Klingl, A.; Pilhofer, M.; Schleper, C. Actin Cytoskeleton and Complex Cell Architecture in an Asgard Archaeon. Nature 2023, 613, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Spring, S.; Bunk, B.; Spröer, C.; Schumann, P.; Rohde, M.; Tindall, B.J.; Klenk, H.-P. Characterization of the First Cultured Representative of Verrucomicrobia Subdivision 5 Indicates the Proposal of a Novel Phylum. ISME J. 2016, 10, 2801–2816. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.-Q.; Han, J.-R.; Zhao, J.-X.; Ye, M.-Q.; Du, Z.-J. Genomic Analysis and Characterization of Pseudotabrizicola Formosa Sp. Nov., a Novel Aerobic Anoxygenic Phototrophic Bacterium, Isolated from Sayram Lake Water. Microorganisms 2022, 10, 2154. [Google Scholar] [CrossRef]
- Katayama, T.; Nobu, M.K.; Kusada, H.; Meng, X.-Y.; Hosogi, N.; Uematsu, K.; Yoshioka, H.; Kamagata, Y.; Tamaki, H. Isolation of a Member of the Candidate Phylum ‘Atribacteria’ Reveals a Unique Cell Membrane Structure. Nat. Commun. 2020, 11, 6381. [Google Scholar] [CrossRef]
- Ibáñez, A.; Barreiro, C.; Diez-Galán, A.; Cobos, R.; Calvo-Peña, C.; Coque, J.J.R. Molecular Identification and Acid Stress Response of an Acidithiobacillus Thiooxidans Strain Isolated from Rio Tinto (Spain). Int. J. Mol. Sci. 2023, 24, 13391. [Google Scholar] [CrossRef]
- Ishii, K.; Fujitani, H.; Soh, K.; Nakagawa, T.; Takahashi, R.; Tsuneda, S. Enrichment and Physiological Characterization of a Cold-Adapted Nitrite-Oxidizing Nitrotoga Sp. from an Eelgrass Sediment. Appl. Environ. Microbiol. 2017, 83, e00549-17. [Google Scholar] [CrossRef]
- Barbosa, R.G.; Van Veelen, H.P.J.; Pinheiro, V.; Sleutels, T.; Verstraete, W.; Boon, N. Enrichment of Hydrogen-Oxidizing Bacteria from High-Temperature and High-Salinity Environments. Appl. Environ. Microbiol. 2021, 87, e02439-20. [Google Scholar] [CrossRef] [PubMed]
- Mendes, I.V.; Garcia, M.B.; Bitencourt, A.C.A.; Santana, R.H.; Lins, P.D.C.; Silveira, R.; Simmons, B.A.; Gladden, J.M.; Kruger, R.H.; Quirino, B.F. Bacterial Diversity Dynamics in Microbial Consortia Selected for Lignin Utilization. PLoS ONE 2021, 16, e0255083. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, X.; Yang, Q.; Chen, Y.; Du, B. Evolution of Microbial Community and Drug Resistance during Enrichment of Tetracycline-Degrading Bacteria. Ecotoxicol. Environ. Saf. 2019, 171, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Yuan, X.; Wang, M.; He, H.; Essengue Samboukel, M.L.P.; Zhang, Y.; Wang, E. Selective Enrichment of Clostridium Spp. by Nutrition Control from Sihe Coal Geological Microbial Communities. Appl. Biochem. Biotechnol. 2020, 192, 952–964. [Google Scholar] [CrossRef]
- Niu, L.; Chen, Y.; Li, Y.; Wang, Y.; Shen, J.; Wang, L.; Zhang, W.; Zhang, H.; Zhao, B. Diversity, Abundance and Distribution Characteristics of Potential Polyethylene and Polypropylene Microplastic Degradation Bacterial Communities in the Urban River. Water Res. 2023, 232, 119704. [Google Scholar] [CrossRef]
- Mu, D.-S.; Liang, Q.-Y.; Wang, X.-M.; Lu, D.-C.; Shi, M.-J.; Chen, G.-J.; Du, Z.-J. Metatranscriptomic and Comparative Genomic Insights into Resuscitation Mechanisms during Enrichment Culturing. Microbiome 2018, 6, 230. [Google Scholar] [CrossRef]
- Kaur, S.; Dhillon, G.S. Recent Trends in Biological Extraction of Chitin from Marine Shell Wastes: A Review. Crit. Rev. Biotechnol. 2015, 35, 44–61. [Google Scholar] [CrossRef]
- López-Mondéjar, R.; Zühlke, D.; Becher, D.; Riedel, K.; Baldrian, P. Cellulose and Hemicellulose Decomposition by Forest Soil Bacteria Proceeds by the Action of Structurally Variable Enzymatic Systems. Sci. Rep. 2016, 6, 25279. [Google Scholar] [CrossRef]
- Sun, Z.-Z.; Ji, B.-W.; Zheng, N.; Wang, M.; Cao, Y.; Wan, L.; Li, Y.-S.; Rong, J.-C.; He, H.-L.; Chen, X.-L.; et al. Phylogenetic Distribution of Polysaccharide-Degrading Enzymes in Marine Bacteria. Front. Microbiol. 2021, 12, 658620. [Google Scholar] [CrossRef]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S Ribosomal DNA Amplification for Phylogenetic Study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A Taxonomically United Database of 16S rRNA Gene Sequences and Whole-Genome Assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Oh, H.-S.; Park, S.-C.; Chun, J. Towards a Taxonomic Coherence between Average Nucleotide Identity and 16S rRNA Gene Sequence Similarity for Species Demarcation of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucl. Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA Gene Sequencing for Species and Strain-Level Microbiome Analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Wang, X.H.; Zhao, Y.; Zhao, X.M.; Yin, H.; Du, Y.G. Screening, Identification and Zymogram Analysis of a Marine Cold-adapted Chitinase-producing Strain. Acta Agric. Boreali-Occident. Sin. 2014, 23, 92–97. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, J.F.; Li, Y.L.; Liu, D.; Wu, R.B.; Liao, B.Q.; Lei, M.; Xiao, X.; Wu, C.L.; He, H.L. Diversity of culturable bacteria and their extracellular active substance properties in alpine lakes of Southwest China. Microbiol. China 2017, 44, 2043–2054. [Google Scholar] [CrossRef]
- Feng, S.; Gao, G.; Zhu, G.W.; Zhang, Y.L.; Qin, B.Q. Bacterial Community Structure in Different Eco-restoration Areas in a Eutrophic Lake Based on 16S rDNA-DGGE and FDC. Chin. J. Appl. Environ. Biol. 2007, 13, 535–540. [Google Scholar]
- Li, R.; Yu, C.; Li, Y.; Lam, T.-W.; Yiu, S.-M.; Kristiansen, K.; Wang, J. SOAP2: An Improved Ultrafast Tool for Short Read Alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A Modular and Extensible Implementation of the RAST Algorithm for Building Custom Annotation Pipelines and Annotating Batches of Genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A Web Server for Prokaryotic Species Circumscription Based on Pairwise Genome Comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S rRNA Gene Sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Cui, Y.-W.; Zhang, H.-Y.; Lu, P.-F.; Peng, Y.-Z. Effects of Carbon Sources on the Enrichment of Halophilic Polyhydroxyalkanoate-Storing Mixed Microbial Culture in an Aerobic Dynamic Feeding Process. Sci. Rep. 2016, 6, 30766. [Google Scholar] [CrossRef]
- Matar, G.; Bilen, M. Culturomics, a Potential Approach Paving the Way toward Bacteriotherapy. Curr. Opin. Microbiol. 2022, 69, 102194. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial Culturomics: Paradigm Shift in the Human Gut Microbiome Study. Clin. Microbiol. Infect. 2012, 18, 1185–1193. [Google Scholar] [CrossRef]
- Stultiens, K.; Van Kessel, M.A.H.J.; Frank, J.; Fischer, P.; Pelzer, C.; Van Alen, T.A.; Kartal, B.; Op Den Camp, H.J.M.; Jetten, M.S.M. Diversity, Enrichment, and Genomic Potential of Anaerobic Methane- and Ammonium-Oxidizing Microorganisms from a Brewery Wastewater Treatment Plant. Appl. Microbiol. Biotechnol. 2020, 104, 7201–7212. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of Ichip for High-Throughput In Situ Cultivation of “Uncultivable” Microbial Species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Gavrish, E.; Bollmann, A.; Epstein, S.; Lewis, K. A Trap for in Situ Cultivation of Filamentous Actinobacteria. J. Microbiol. Methods 2008, 72, 257–262. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, A.; Crawford, J.M.; Stewart, E.J.; Witt, K.; Gavrish, E.; Epstein, S.; Clardy, J.; Lewis, K. Siderophores from Neighboring Organisms Promote the Growth of Uncultured Bacteria. Chem. Biol. 2010, 17, 254–264. [Google Scholar] [CrossRef]
- McCully, A.L.; Loop Yao, M.; Brower, K.K.; Fordyce, P.M.; Spormann, A.M. Double Emulsions as a High-Throughput Enrichment and Isolation Platform for Slower-Growing Microbes. ISME Commun. 2023, 3, 47. [Google Scholar] [CrossRef]
- Kharade, S.S.; McBride, M.J. Flavobacterium Johnsoniae Chitinase ChiA Is Required for Chitin Utilization and Is Secreted by the Type IX Secretion System. J. Bacteriol. 2014, 196, 961–970. [Google Scholar] [CrossRef]
- Midorikawa, Y.; Shimizu, T.; Sanda, T.; Hamasaki, K.; Dan, S.; Lal, M.T.B.M.; Kato, G.; Sano, M. Characterization of Aquimarina Hainanensis Isolated from Diseased Mud Crab Scylla Serrata Larvae in a Hatchery. J. Fish. Dis. 2020, 43, 541–549. [Google Scholar] [CrossRef]
- Daniels, M.; Van Vliet, S.; Ackermann, M. Changes in Interactions over Ecological Time Scales Influence Single-Cell Growth Dynamics in a Metabolically Coupled Marine Microbial Community. ISME J. 2023, 17, 406–416. [Google Scholar] [CrossRef]
- Liu, J.; Cai, S.-D.; Wang, Y.-J.; Wang, Q.; Zhang, D.-C. Algibacter Pacificus Sp. Nov., Isolated from a Deep-Sea Seamount. Int. J. Syst. Evol. Microbiol. 2020, 70, 2907–2911. [Google Scholar] [CrossRef]
- Cortes-Tolalpa, L. Halotolerant Microbial Consortia Able to Degrade Highly Recalcitrant Plant Biomass Substrate. Appl. Microbiol. Biotechnol. 2018, 102, 2913–2927. [Google Scholar] [CrossRef]
- Gavande, P.V.; Basak, A.; Sen, S.; Lepcha, K.; Murmu, N.; Rai, V.; Mazumdar, D.; Saha, S.P.; Das, V.; Ghosh, S. Functional Characterization of Thermotolerant Microbial Consortium for Lignocellulolytic Enzymes with Central Role of Firmicutes in Rice Straw Depolymerization. Sci. Rep. 2021, 11, 3032. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-H.; Lee, M.-H.; Jung, Y.-T. Pseudofulvibacter Geojedonensis Gen. Nov., Sp. Nov., a Polysaccharide-Degrading Member of the Family Flavobacteriaceae Isolated from Seawater, and Emended Description of the Genus Fulvibacter. Int. J. Syst. Evol. Microbiol. 2013, 63, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.; Lee, Y.M.; Hwang, C.Y.; Park, H.; Jung, Y.-J.; Kim, M.-K.; Hong, S.G.; Kim, J.H.; Lee, H.K. Psychroserpens Jangbogonensis Sp. Nov., a Psychrophilic Bacterium Isolated from Antarctic Marine Sediment. Int. J. Syst. Evol. Microbiol. 2015, 65, 183–188. [Google Scholar] [CrossRef]
- Lin, H.; Lin, D.; Zhang, M.; Ye, J.; Sun, J.; Tang, K. Maribacter Hydrothermalis Sp. Nov., Isolated from Shallow-Sea Hydrothermal Systems Off Kueishantao Island. Curr. Microbiol. 2021, 78, 2815–2820. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, D.M.; Palakawong Na Ayudthaya, S.; Diop, S.; Villanueva, L.; Stams, A.J.M.; Sánchez-Andrea, I. Anaerobic Degradation of Sulfated Polysaccharides by Two Novel Kiritimatiellales Strains Isolated From Black Sea Sediment. Front. Microbiol. 2019, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, H.; Ushiki, N.; Tsuneda, S.; Aoi, Y. Isolation of Sublineage I Nitrospira by a Novel Cultivation Strategy: Isolation of Uncultured Nitrospira. Environ. Microbiol. 2014, 16, 3030–3040. [Google Scholar] [CrossRef]
- Hu, H.; Natarajan, V.P.; Wang, F. Towards Enriching and Isolation of Uncultivated Archaea from Marine Sediments Using a Refined Combination of Conventional Microbial Cultivation Methods. Mar. Life Sci. Technol. 2021, 3, 231–242. [Google Scholar] [CrossRef]
- Grondin, J.M.; Tamura, K.; Déjean, G.; Abbott, D.W.; Brumer, H. Polysaccharide Utilization Loci: Fueling Microbial Communities. J. Bacteriol. 2017, 199, 426. [Google Scholar] [CrossRef]
- McKee, L.S.; La Rosa, S.L.; Westereng, B.; Eijsink, V.G.; Pope, P.B.; Larsbrink, J. Polysaccharide Degradation by the Bacteroidetes: Mechanisms and Nomenclature. Environ. Microbiol. Rep. 2021, 13, 559–581. [Google Scholar] [CrossRef]
- Munoz, R.; Rosselló-Móra, R.; Amann, R. Revised Phylogeny of Bacteroidetes and Proposal of Sixteen New Taxa and Two New Combinations Including Rhodothermaeota Phyl. Nov. Syst. Appl. Microbiol. 2016, 39, 281–296. [Google Scholar] [CrossRef]
- Hahnke, R.L.; Meier-Kolthoff, J.P.; García-López, M.; Mukherjee, S.; Huntemann, M.; Ivanova, N.N.; Woyke, T.; Kyrpides, N.C.; Klenk, H.-P.; Göker, M. Genome-Based Taxonomic Classification of Bacteroidetes. Front. Microbiol. 2016, 7, 2003. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Muntyan, M.S.; Toshchakov, S.V.; Korzhenkov, A.; Kublanov, I.V. Phenotypic and Genomic Properties of a Novel Deep-Lineage Haloalkaliphilic Member of the Phylum Balneolaeota From Soda Lakes Possessing Na+-Translocating Proteorhodopsin. Front. Microbiol. 2018, 9, 2672. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Liu, J.J.; Yu, Z.H.; Wang, X.Z.; Jin, J.; Liu, X.B. Research Progress of Acidobacteria Ecology in Soils. Biotechnol. Bull. 2016, 32, 14–20. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative Metagenomic, Phylogenetic and Physiological Analyses of Soil Microbial Communities across Nitrogen Gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, K.S.; Craine, J.M.; Fierer, N. Consistent Effects of Nitrogen Amendments on Soil Microbial Communities and Processes across Biomes. Glob. Chang. Biol. 2012, 18, 1918–1927. [Google Scholar] [CrossRef]
- Sichert, A. Verrucomicrobia Use Hundreds of Enzymes to Digest the Algal Polysaccharide Fucoidan. Nat. Microbiol. 2020, 5, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Matsuo, Y.; Adachi, K.; Nozawa, M.; Matsuda, S.; Kasai, H.; Yokota, A. Description of Persicirhabdus Sediminis Gen. Nov., Sp. Nov., Roseibacillus Ishigakijimensis Gen. Nov., Sp. Nov., Roseibacillus Ponti Sp. Nov., Roseibacillus Persicicus Sp. Nov., Luteolibacter Pohnpeiensis Gen. Nov., Sp. Nov. and Luteolibacter Algae Sp. Nov., Six Marine Members of the Phylum “Verrucomicrobia”, and Emended Descriptions of the Class Verrucomicrobiae, the Order Verrucomicrobiales and the Family Verrucomicrobiaceae. Int. J. Syst. Evol. Microbiol. 2008, 58, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Matsuo, Y.; Katsuta, A.; Jang, J.-H.; Matsuda, S.; Adachi, K.; Kasai, H.; Yokota, A. Haloferula rosea gen. nov., sp. nov., Haloferula harenae sp. nov., Haloferula phyci sp. nov., Haloferula helveola sp. nov. and Haloferula sargassicola sp. nov., Five Marine Representatives of the Family Verrucomicrobiaceae within the Phylum “Verrucomicrobia”. Int. J. Syst. Evol. Microbiol. 2008, 58, 2491–2500. [Google Scholar] [CrossRef]
- Zhang, C.-J.; Chen, Y.-L.; Sun, Y.-H.; Pan, J.; Cai, M.-W.; Li, M. Diversity, Metabolism and Cultivation of Archaea in Mangrove Ecosystems. Mar. Life Sci. Technol. 2021, 3, 252–262. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strains | Size (bp) | N50 Len. (bp) | GC Content (%) | Number of Coding Sequences | Genes Related to the Metabolism of Cellulose and Chitin and Their Degradation Products |
|---|---|---|---|---|---|
| 4AJ11 | 3,177,342 | 1,663,422 | 49.3 | 2881 | beta-N-acetylglucosaminidase (EC 3.2.1.52) |
| 28AX09 | 3,151,738 | 1,843,572 | 56.9 | 2906 | endoglucanase, beta-glucosidase (EC 3.2.1.21), beta-N-acetylglucosaminidase (EC 3.2.1.52), beta-glycosyl hydrolase |
| 57AJ16 | 4,920,416 | 227,616 | 34.3 | 4385 | endoglucanase beta-glucosidase (EC 3.2.1.21) chitinase (EC 3.2.1.14) beta-glycosyl hydrolase N-acetylmuramic acid 6-phosphate etherase (EC 4.2.1.126) N-acetyl glucosamine transporter (NagP) N-acetylglucosamine related transporter (NagX) Glucosamine-6-phosphate deaminase (EC 3.5.99.6) beta-glycosyl hydrolase Glucosamine-6-phosphate deaminase (EC 3.5.99.6) |
| 57CJ19 | 3,915,335 | 2,086,907 | 61.2 | 3913 | beta-glucosidase (EC 3.2.1.21) beta-N-acetylglucosaminidase (EC 3.2.1.52) N-acetylmuramic acid 6-phosphate etherase (EC 4.2.1.126) Glucosamine-6-phosphate deaminase (EC 3.5.99.6) N-acetylglucosamine kinase of eukaryotic type (EC 2.7.1.59) N-acetylglucosamine-6-phosphate deacetylase (EC 3.5.1.25) Glucosamine-6-phosphate deaminase (EC 3.5.99.6) |
| 92AX17 | 3,864,317 | 1,137,868 | 66.6 | 3809 | beta-glucosidase (EC 3.2.1.21) beta-N-acetylglucosaminidase (EC 3.2.1.52) Glucosamine-6-phosphate deaminase (EC 3.5.99.6) N-acetylglucosamine-6-phosphate deacetylase (EC 3.5.1.25) Glucosamine-6-phosphate deaminase (EC 3.5.99.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Zheng, N.; Li, X.; Zhao, K.; Xie, B.-B. Enrichment Pretreatment Expands the Microbial Diversity Cultivated from Marine Sediments. Microorganisms 2023, 11, 2771. https://doi.org/10.3390/microorganisms11112771
Wang M, Zheng N, Li X, Zhao K, Xie B-B. Enrichment Pretreatment Expands the Microbial Diversity Cultivated from Marine Sediments. Microorganisms. 2023; 11(11):2771. https://doi.org/10.3390/microorganisms11112771
Chicago/Turabian StyleWang, Meng, Ning Zheng, Xuan Li, Kun Zhao, and Bin-Bin Xie. 2023. "Enrichment Pretreatment Expands the Microbial Diversity Cultivated from Marine Sediments" Microorganisms 11, no. 11: 2771. https://doi.org/10.3390/microorganisms11112771
APA StyleWang, M., Zheng, N., Li, X., Zhao, K., & Xie, B.-B. (2023). Enrichment Pretreatment Expands the Microbial Diversity Cultivated from Marine Sediments. Microorganisms, 11(11), 2771. https://doi.org/10.3390/microorganisms11112771