
Tracking the Selective Pressure Profile and Gene Flow of SARS-CoV-2 Delta Variant in Italy from April to October 2021 and Frequencies of Key Mutations from Three Representative Italian Regions

 , , , , ,
, , , , ,  , , , , , , ,
, , , , , , ,  ,
,  , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset and Sequence Alignment
2.2. Gene Flow and Migration Analysis
2.3. Selective Pressure Analysis
3. Results
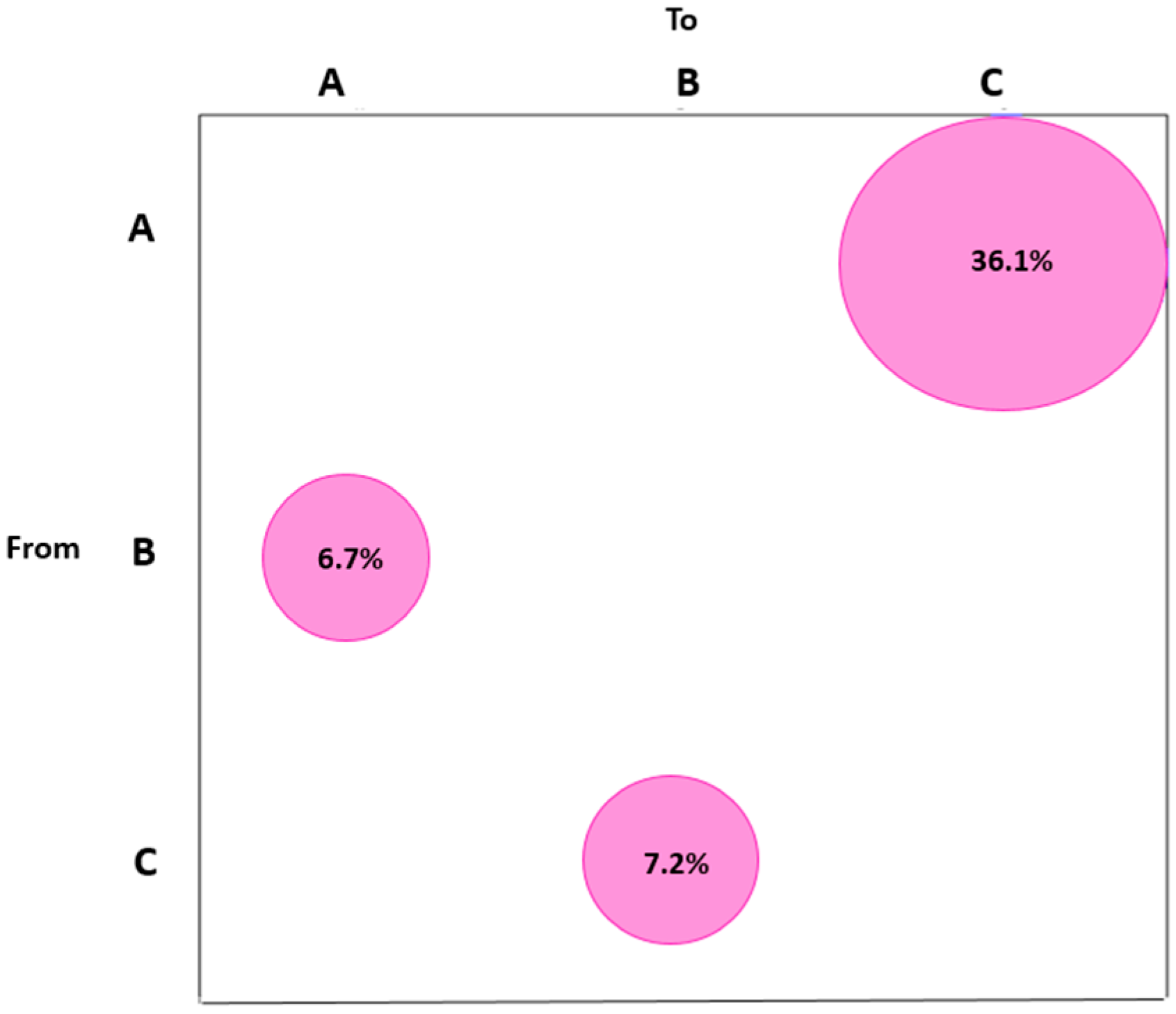
Gene Flow and Selective Pressure Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Centre for Disease Prevention and Control (ECDC). SARS-CoV-2 Variants of Concern as of 23 March 2023. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 5 October 2022).
- Mahilkar, S.; Agrawal, S.; Chaudhary, S.; Parikh, S.; Sonkar, S.C.; Verma, D.K.; Chitalia, V.; Mehta, D.; Koner, B.C.; Vijay, N.; et al. SARS-CoV-2 variants: Impact on biological and clinical outcome. Front. Med. 2022, 9, 995960. [Google Scholar] [CrossRef]
- Cov-Lineages.org–Lineage List. Available online: https://cov-lineages.org/lineage_list.html (accessed on 5 October 2022).
- Stima Della Prevalenza delle Varianti VOC (Variants of Concern) in Italia: Beta, Gamma, Delta, Omicron e Altre Varianti di SARS-CoV-2. Quick Survey 20 December 2021. Available online: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-indagini-rapide-20-dicembre-2021.pdf (accessed on 5 October 2022).
- Prevalenza e Distribuzione Delle Varianti di SARS-CoV-2 di Interesse per la Sanità Pubblica in Italia Rapporto n. 15–10 December 2021. Available online: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-rapporti-periodici-10-dicembre-2021.pdf (accessed on 5 October 2022).
- European Centre for Disease Prevention and Control. Communicable Disease Threats Report, Week 32 7–13 August 2022. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/Communicable-disease-threats-report-13-aug-2022-all-users.pdf (accessed on 10 October 2022).
- European Centre for Disease Prevention and Control. ECDC de-Escalates BA.2, BA.4 and BA.5 from Its List of Variants of Concern. Available online: https://www.ecdc.europa.eu/en/news-events/ecdc-de-escalates-ba2-ba4-and-ba5-its-list-variants-concern (accessed on 3 March 2023).
- Middleton, C.; Kubatko, L. Assessment of positive selection across SARS-CoV-2 variants via maximum likelihood. PLoS ONE 2023, 18, e0291271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, L.; Xu, H.; Fu, Y.; Peng, X.; Zheng, Y.; Yu, J.; He, J. Evolutionary Pattern Comparisons of the SARS-CoV-2 Delta Variant in Countries/Regions with High and Low Vaccine Coverage. Viruses 2022, 14, 2296. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Melnychuk, S.; Sandstrom, P.; Ji, H. Tracking the evolution of the SARS-CoV-2 Delta variant of concern: Analysis of genetic diversity and selection across the whole viral genome. Front. Microbiol. 2023, 14, 1222301. [Google Scholar] [CrossRef]
- De Marco, C.; Veneziano, C.; Massacci, A.; Pallocca, M.; Marascio, N.; Quirino, A.; Barreca, G.S.; Giancotti, A.; Gallo, L.; Lamberti, A.G.; et al. Dynamics of Viral Infection and Evolution of SARS-CoV-2 Variants in the Calabria Area of Southern Italy. Front. Microbiol. 2022, 13, 934993. [Google Scholar] [CrossRef]
- Baj, A.; Novazzi, F.; Ferrante, F.D.; Genoni, A.; Tettamanzi, E.; Catanoso, G. Spike protein evolution in the SARS-CoV-2 Delta variant of concern: A case series from Northern Lombardy. Emerg. Microbes Infect. 2021, 10, 2010–2015. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Della Ventura, C.; Menzo, S.; Bruzzone, B.; Sagradi, F.; Ceccherini-Silberstein, F.; Weisz, A.; Clementi, N.; Brindicci, G.; et al. Epidemiological and Clinical Features of SARS-CoV-2 Variants Circulating between April–December 2021 in Italy. Viruses 2022, 14, 2508. [Google Scholar] [CrossRef]
- Petrone, D.; Mateo-Urdiales, A.; Sacco, C.; Riccardo, F.; Bella, A.; Ambrosio, L.; Presti, A.L.; Di Martino, A.; Ceccarelli, E.; Del Manso, M.; et al. Reduction of the risk of severe COVID-19 due to Omicron compared to Delta variant in Italy (November 2021–February 2022). Int. J. Infect. Dis. 2023, 129, 135–141. [Google Scholar] [CrossRef]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positive selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. A genetic algorithm approach to detecting lineage-specific variation in selection pressure. Mol. Biol. Evol. 2005, 22, 478–485. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Public Health Impact of SARS-CoV-2 Variants of Concern: Scoping Review Protocol. Available online: https://www.ecdc.europa.eu/en/publications-data/public-health-impact-sars-cov-2-variants-concern-scoping-review-protocol (accessed on 18 May 2021).
- GISAID. Available online: https://gisaid.org/ (accessed on 14 October 2021).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Galaxy Platform. Available online: https://usegalaxy.org/ (accessed on 21 October 2021).
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Slatkin, M.; Maddison, W.P. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 1989, 123, 603–613. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not So Different after All: A Comparison of Methods for Detecting Amino Acid Sites under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W.; Spencer, V.M. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef]
- Ghosh, N.; Nandi, S.; Saha, I. Phylogenetic analysis of 17271 Indian SARS-CoV-2 genomes to identify temporal and spatial hotspot mutations. PLoS ONE 2022, 17, e0265579. [Google Scholar] [CrossRef]
- WHO. Weekly Epidemiological Update on COVID-19-30 March 2021. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---31-march-2021 (accessed on 20 October 2022).
- Tian, D.; Sun, Y.; Zhou, J.; Ye, Q. The Global Epidemic of the SARS-CoV-2 Delta Variant, Key Spike Mutations and Immune Escape. Front. Immunol. 2021, 12, 751778. [Google Scholar] [CrossRef]
- Chavda, V.P.; Bezbaruah, R.; Deka, K.; Nongrang, L.; Kalita, T. The Delta and Omicron Variants of SARS-CoV-2: What We Know So Far. Vaccines 2022, 10, 1926. [Google Scholar] [CrossRef]
- Stima della Prevalenza delle Varianti VOC (Variant Of Concern) e di altre varianti di SARS-CoV-2 in Italia.Quick Survey 17 January 2022. Available online: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-indagini-rapide-17-gennaio-2022.pdf (accessed on 20 October 2022).
- Stima Della Prevalenza Delle Varianti VOC (Variant of Concern) e di Altre Varianti di SARS-CoV-2 in Italia. Quick Survey 31 January 2022. Available online: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-indagini-rapide-31-gennaio-2022.pdf (accessed on 20 October 2022).
- Véras, N.M.C.; Santoro, M.M.; Gray, R.R.; Tatem, A.J.; Presti, A.L.; Olearo, F.; Cappelli, G.; Colizzi, V.; Takou, D.; Torimiro, J.; et al. Molecular epidemiology of HIV type 1 CRF02_AG in Cameroon and African patients living in Italy. AIDS Res. Hum. Retrovir. 2011, 27, 1173–1182. [Google Scholar] [CrossRef]
- Lo Presti, A.; Rezza, G.; Stefanelli, P. Selective pressure on SARS-CoV-2 protein coding genes and glycosylation site prediction. Heliyon 2020, 6, e05001. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Sharma, K.; Chand, H.S.; Byrareddy, S.N.; Singh, K. Omicron SARS-CoV-2 variant: Unique features and their impact on pre-existing antibodies. J. Autoimmun. 2022, 126, 102779. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.K.; Kumar, S.; Ansari, S.; Paweska, J.T.; Maurya, V.K.; Tripathi, A.K.; Abdel-Moneim, A.S. Characterization of the novel SARS-CoV-2 Omicron (B.1.1.529) variant of concern and its global perspective. J. Med. Virol. 2022, 94, 1738–1744. [Google Scholar] [CrossRef] [PubMed]
- Magazine, N.; Zhang, T.; Wu, Y.; McGee, M.C.; Veggiani, G.; Huang, W. Mutations and Evolution of the SARS-CoV-2 Spike Protein. Viruses 2022, 14, 640. [Google Scholar] [CrossRef]
- Upadhyay, V.; Lucas, A.; Panja, S.; Miyauchi, R.; Mallela, K.M.G. Receptor binding, immune escape, and protein stability direct the natural selection of SARS-CoV-2 variants. J. Biol. Chem. 2021, 297, 101208. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477-488.e4. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332-2347.e16. [Google Scholar] [CrossRef]
- Shen, L.; Triche, T.J.; Bard, J.D.; Biegel, J.A.; Judkins, A.R.; Gai, X. Spike Protein NTD mutation G142D in SARS-CoV-2 Delta VOC lineages is associated with frequent back mutations, increased viral loads, and immune evasion. medRxiv 2021, 12, 21263475. [Google Scholar] [CrossRef]
- COG. UK-UK Data. Available online: https://sars2.cvr.gla.ac.uk/cog-uk/ (accessed on 17 May 2023).

{kind=link}
| Mutation | Target | % (n = 1500) | % (n = 596) | % (n = 213, Lazio) | % (n = 245, Sicily) | % (n = 138, Veneto) |
|---|---|---|---|---|---|---|
| P62S | nsp1 | 0.50% | 0.70% | 1.40% | 0.40% | 0.00% |
| E87D | nsp1 | 4.20% | 5.20% | 7.00% | 5.30% | 2.20% |
| G94S | nsp1 | 0.50% | 1.20% | 2.80% | 0.00% | 0.70% |
| G94V | nsp1 | 0.10% | 0.20% | 0.00% | 0.40% | 0.00% |
| R27C | nsp2 | 1.70% | 0.00% | 0.00% | 0.00% | 0.00% |
| K81N | nsp2 | 15.20% | 24.20% | 15.00% | 36.70% | 15.90% |
| E89K | nsp2 | 0.90% | 2.30% | 0.00% | 5.70% | 0.00% |
| P129L | nsp2 | 4.70% | 3.40% | 2.80% | 2.90% | 5.10% |
| P129S | nsp2 | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| D155G | nsp2 | 0.90% | 0.80% | 1.40% | 0.80% | 0.00% |
| A159V | nsp2 | 0.30% | 0.00% | 0.00% | 0.00% | 0.00% |
| S263F | nsp2 | 3.10% | 6.00% | 1.40% | 13.50% | 0.00% |
| A318V | nsp2 | 6.10% | 11.90% | 4.20% | 21.60% | 6.50% |
| G339S | nsp2 | 0.80% | 1.30% | 0.90% | 1.60% | 1.40% |
| V485I | nsp2 | 2.30% | 1.30% | 1.40% | 0.80% | 2.20% |
| Q496P | nsp2 | 1.20% | 0.80% | 0.90% | 1.20% | 0.00% |
| Q496H | nsp2 | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| S126L | nsp3 | 0.30% | 0.20% | 0.00% | 0.00% | 0.70% |
| K384N | nsp3 | 0.30% | 0.30% | 0.50% | 0.00% | 0.70% |
| L862F | nsp3 | 0.20% | 0.00% | 0.00% | 0.00% | 0.00% |
| P1228L | nsp3 | 74.10% | 78.20% | 88.30% | 66.50% | 83.30% |
| L1791F | nsp3 | 0.30% | 0.20% | 0.00% | 0.00% | 0.70% |
| T204I | nsp4 | 0.40% | 0.70% | 0.90% | 0.40% | 0.70% |
| D279N | nsp4 | 0.20% | 0.00% | 0.00% | 0.00% | 0.00% |
| T295I | nsp4 | 2.20% | 5.00% | 0.00% | 12.20% | 0.00% |
| C296F | nsp4 | 1.30% | 0.70% | 0.90% | 0.40% | 0.70% |
| A2V | nsp6 | 2.60% | 2.30% | 3.30% | 0.00% | 5.10% |
| T6I | nsp6 | 0.50% | 1.20% | 3.30% | 0.00% | 0.00% |
| Q27R | nsp6 | 0.50% | 1.20% | 0.00% | 0.00% | 5.10% |
| L37F | nsp6 | 2.10% | 1.80% | 4.20% | 0.40% | 0.70% |
| A46S | nsp12 | 2.30% | 5.20% | 0.00% | 12.70% | 0.00% |
| E61D | nsp12 | 1.40% | 3.50% | 0.00% | 8.60% | 0.00% |
| E61K | nsp12 | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| A95S | nsp12 | 0.50% | 0.30% | 0.90% | 0.00% | 0.00% |
| T141I | nsp12 | 0.50% | 0.00% | 0.00% | 0.00% | 0.00% |
| R197Q | nsp12 | 2.30% | 3.00% | 3.30% | 0.80% | 6.50% |
| P323L | nsp12 | 98.50% | 97.30% | 100.00% | 93.50% | 100.00% |
| S384P | nsp12 | 1.10% | 0.80% | 0.90% | 1.20% | 0.00% |
| M463I | nsp12 | 1.00% | 0.80% | 0.50% | 1.60% | 0.00% |
| Q822H | nsp12 | 3.10% | 5.70% | 13.60% | 0.40% | 2.90% |
| L838I | nsp12 | 12.80% | 12.40% | 16.00% | 4.10% | 21.70% |
| P77L | nsp13 | 95.90% | 99.50% | 100.00% | 100.00% | 97.80% |
| V89I | nsp13 | 0.30% | 0.00% | 0.00% | 0.00% | 0.00% |
| P46L | nsp14 | 2.10% | 2.20% | 1.90% | 2.90% | 1.40% |
| R289H | nsp14 | 3.00% | 5.20% | 12.70% | 0.80% | 1.40% |
| S374F | nsp14 | 0.40% | 0.30% | 0.50% | 0.00% | 0.70% |
| A394V | nsp14 | 69.00% | 78.20% | 88.30% | 66.50% | 83.30% |
| A80V | nsp15 | 0.50% | 1.00% | 0.00% | 2.40% | 0.00% |
| A81V | nsp15 | 0.70% | 0.20% | 0.50% | 0.00% | 0.00% |
| V9I | nsp16 | 0.40% | 0.00% | 0.00% | 0.00% | 0.00% |
| A34V | nsp16 | 0.50% | 1.20% | 0.00% | 1.60% | 2.20% |
| P215L | nsp16 | 0.10% | 0.20% | 0.50% | 0.00% | 0.00% |
| P215T | nsp16 | 0.70% | 0.80% | 0.90% | 1.20% | 0.00% |
| L5F | spike | 0.90% | 0.30% | 0.50% | 0.00% | 0.70% |
| V70I | spike | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| V70F | spike | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| T95I | spike | 20.90% | 16.80% | 19.70% | 11.80% | 21.00% |
| G142D | spike | 64.10% | 73.50% | 55.40% | 93.10% | 66.70% |
| G142Y | spike | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| G142H | spike | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| G142V | spike | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| A222V | spike | 20.90% | 19.00% | 8.90% | 30.60% | 13.80% |
| A222S | spike | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| V367L | spike | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| V367H | spike | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| V367F | spike | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| L452R | spike | 98.60% | 98.00% | 94.40% | 100.00% | 100.00% |
| Q613H | spike | 6.60% | 13.60% | 1.90% | 29.80% | 2.90% |
| N501Y | spike | 1.20% | 0.20% | 0.00% | 0.40% | 0.00% |
| D614G | spike | 90.80% | 100.00% | 100.00% | 100.00% | 100.00% |
| Q677H | spike | 3.80% | 4.00% | 5.60% | 1.60% | 5.80% |
| P681R | spike | 93.90% | 100.00% | 100.00% | 100.00% | 100.00% |
| D950N | spike | 83.70% | 82.00% | 91.50% | 65.30% | 97.10% |
| V1104L | spike | 0.90% | 0.70% | 0.50% | 0.40% | 1.40% |
| V1128L | spike | 1.70% | 0.00% | 0.00% | 0.00% | 0.00% |
| G1219V | spike | 0.40% | 0.20% | 0.00% | 0.40% | 0.00% |
| G1219C | spike | 0.20% | 0.00% | 0.00% | 0.00% | 0.00% |
| L41F | ORF3a | 0.30% | 0.30% | 0.50% | 0.40% | 0.00% |
| L41I | ORF3a | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| A110S | ORF3a | 2.90% | 6.00% | 0.50% | 14.30% | 0.00% |
| A110V | ORF3a | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
| I82T | M | 2.00% | 100.00% | 100.00% | 100.00% | 100.00% |
| Q9L | N | 14.10% | 12.10% | 16.00% | 4.10% | 20.30% |
| Q9H | N | 0.10% | 0.00% | 0.00% | 0.00% | 0.00% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Presti, A.; Di Martino, A.; Ambrosio, L.; De Sabato, L.; Knijn, A.; Vaccari, G.; Di Bartolo, I.; Morabito, S.; Terregino, C.; Fusaro, A.; et al. Tracking the Selective Pressure Profile and Gene Flow of SARS-CoV-2 Delta Variant in Italy from April to October 2021 and Frequencies of Key Mutations from Three Representative Italian Regions. Microorganisms 2023, 11, 2644. https://doi.org/10.3390/microorganisms11112644
Lo Presti A, Di Martino A, Ambrosio L, De Sabato L, Knijn A, Vaccari G, Di Bartolo I, Morabito S, Terregino C, Fusaro A, et al. Tracking the Selective Pressure Profile and Gene Flow of SARS-CoV-2 Delta Variant in Italy from April to October 2021 and Frequencies of Key Mutations from Three Representative Italian Regions. Microorganisms. 2023; 11(11):2644. https://doi.org/10.3390/microorganisms11112644
Chicago/Turabian StyleLo Presti, Alessandra, Angela Di Martino, Luigina Ambrosio, Luca De Sabato, Arnold Knijn, Gabriele Vaccari, Ilaria Di Bartolo, Stefano Morabito, Calogero Terregino, Alice Fusaro, and et al. 2023. "Tracking the Selective Pressure Profile and Gene Flow of SARS-CoV-2 Delta Variant in Italy from April to October 2021 and Frequencies of Key Mutations from Three Representative Italian Regions" Microorganisms 11, no. 11: 2644. https://doi.org/10.3390/microorganisms11112644
APA StyleLo Presti, A., Di Martino, A., Ambrosio, L., De Sabato, L., Knijn, A., Vaccari, G., Di Bartolo, I., Morabito, S., Terregino, C., Fusaro, A., Monne, I., Giussani, E., Tramuto, F., Maida, C. M., Mazzucco, W., Costantino, C., Rueca, M., Giombini, E., Gruber, C. E. M., ... on behalf of the Italian Genomic Laboratory Network. (2023). Tracking the Selective Pressure Profile and Gene Flow of SARS-CoV-2 Delta Variant in Italy from April to October 2021 and Frequencies of Key Mutations from Three Representative Italian Regions. Microorganisms, 11(11), 2644. https://doi.org/10.3390/microorganisms11112644