
Unlocking the Viral Universe: Metagenomic Analysis of Bat Samples Using Next-Generation Sequencing

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Samples Preparation and Metagenomic Sequencing
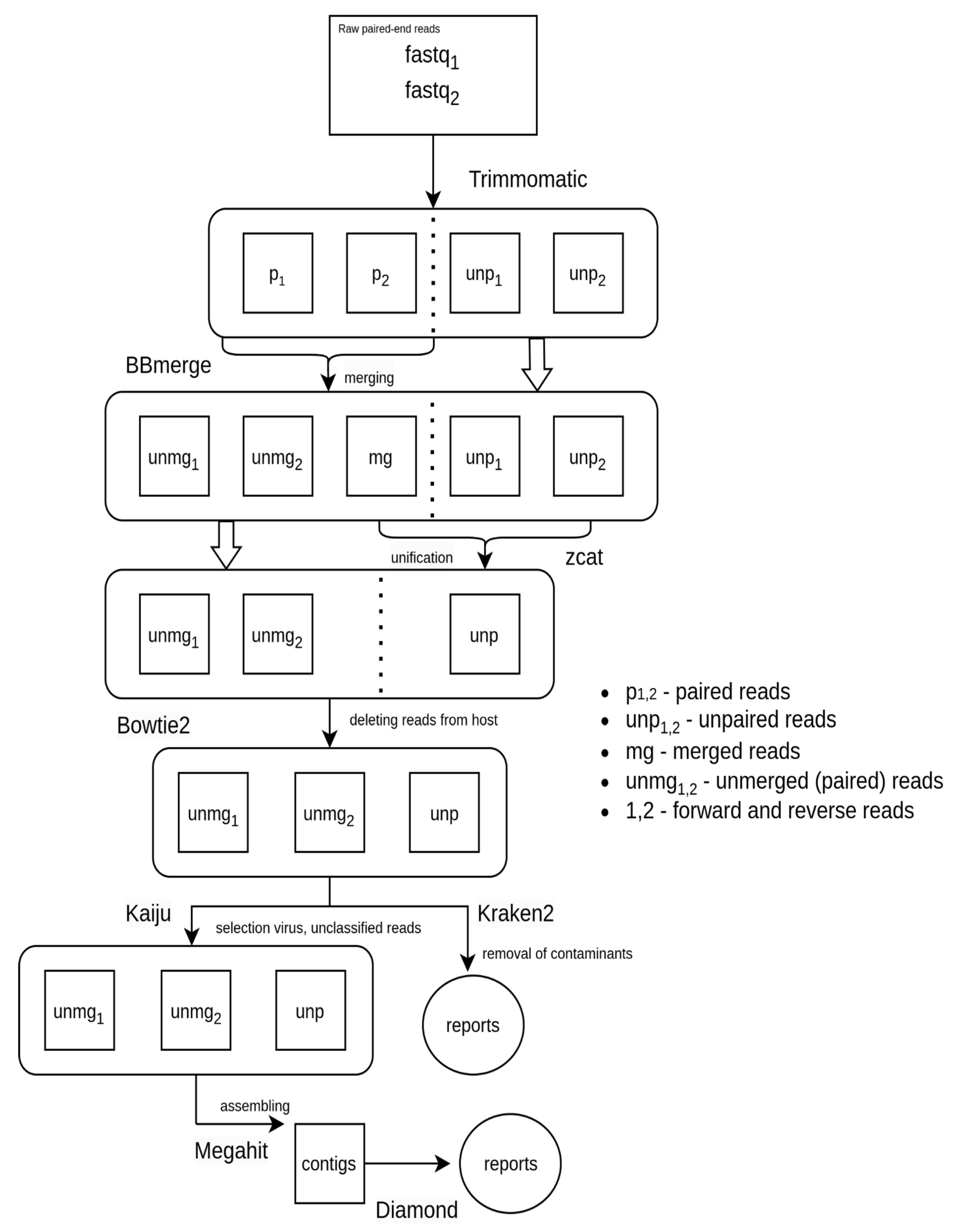
2.3. Bioinformatics Analysis of Metagenomic Sequencing Data
2.4. Phylogenetic Analysis
2.5. Data Processing after SMART
3. Results
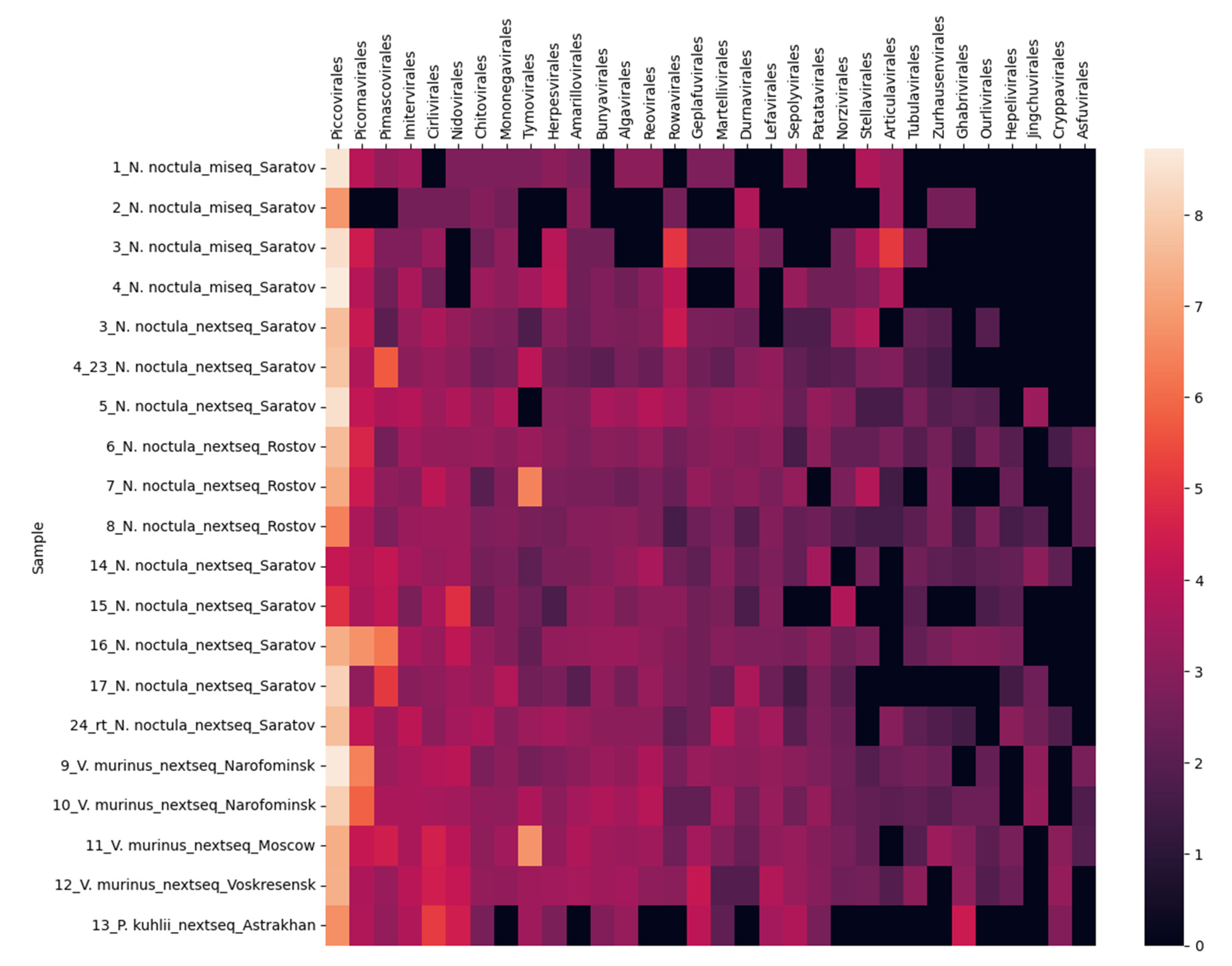
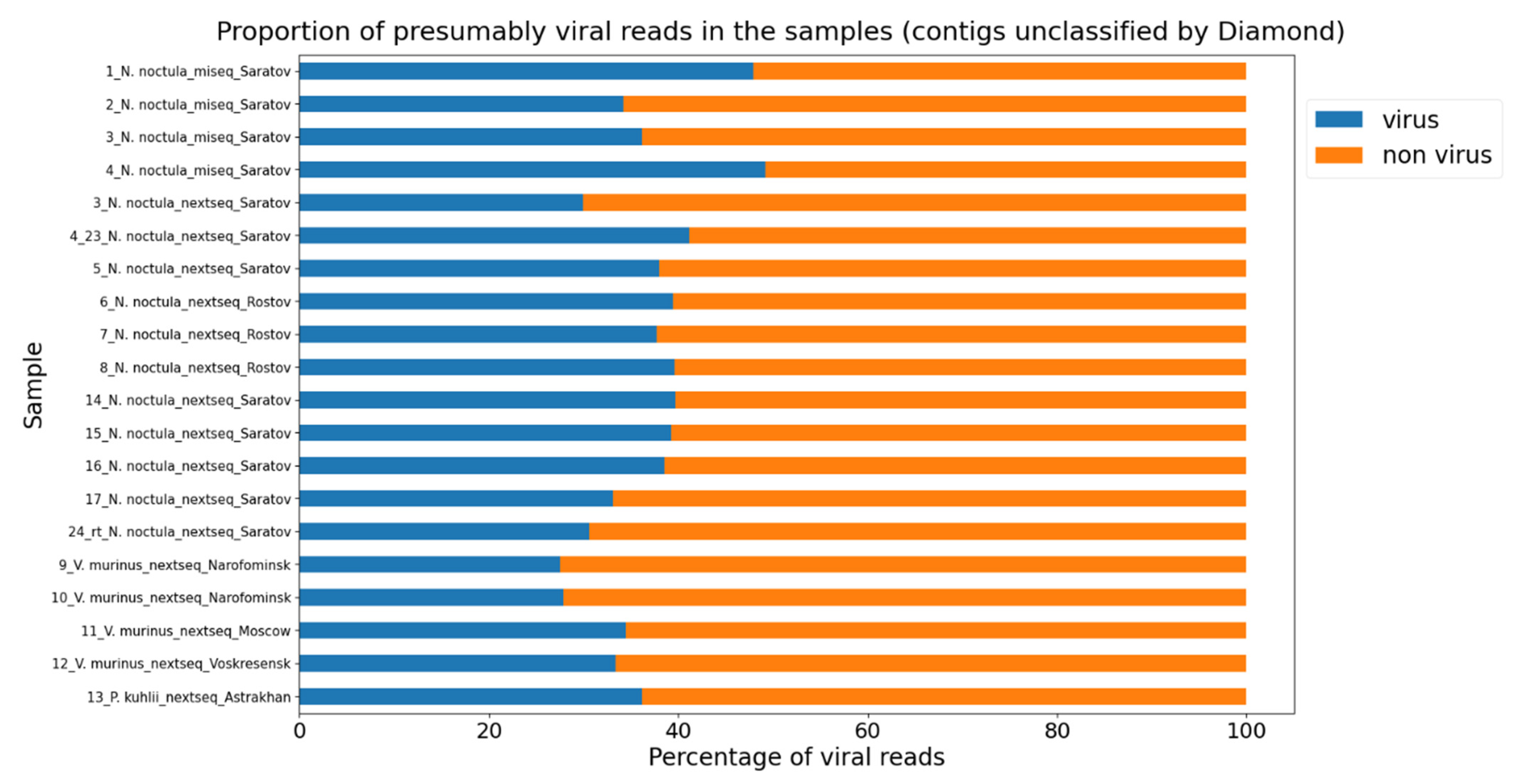
3.1. Metagenomic Sequencing of Bat Fecal Samples
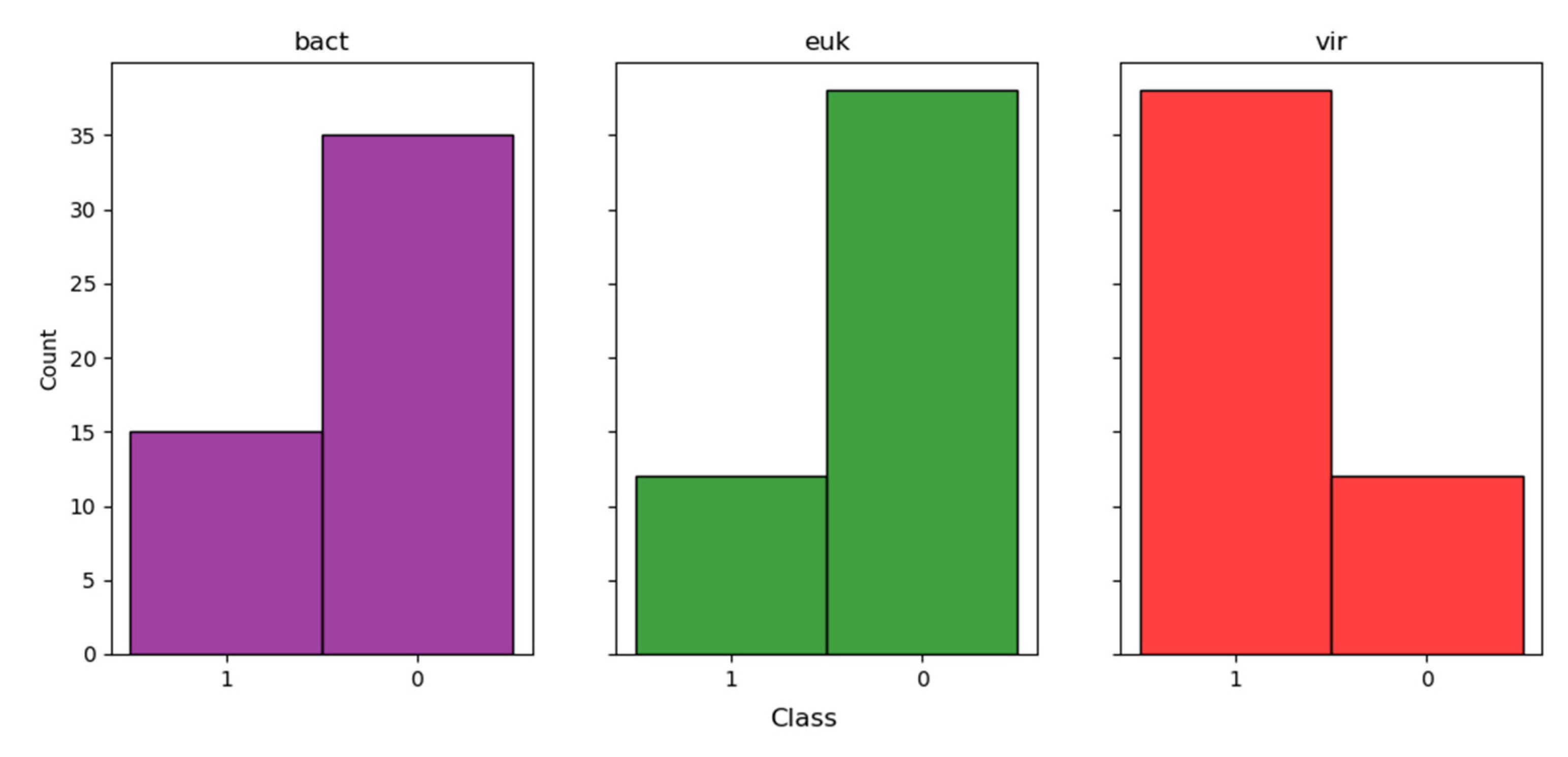
3.2. A Neural Network Approach for Finding Presumptive Viral Reads
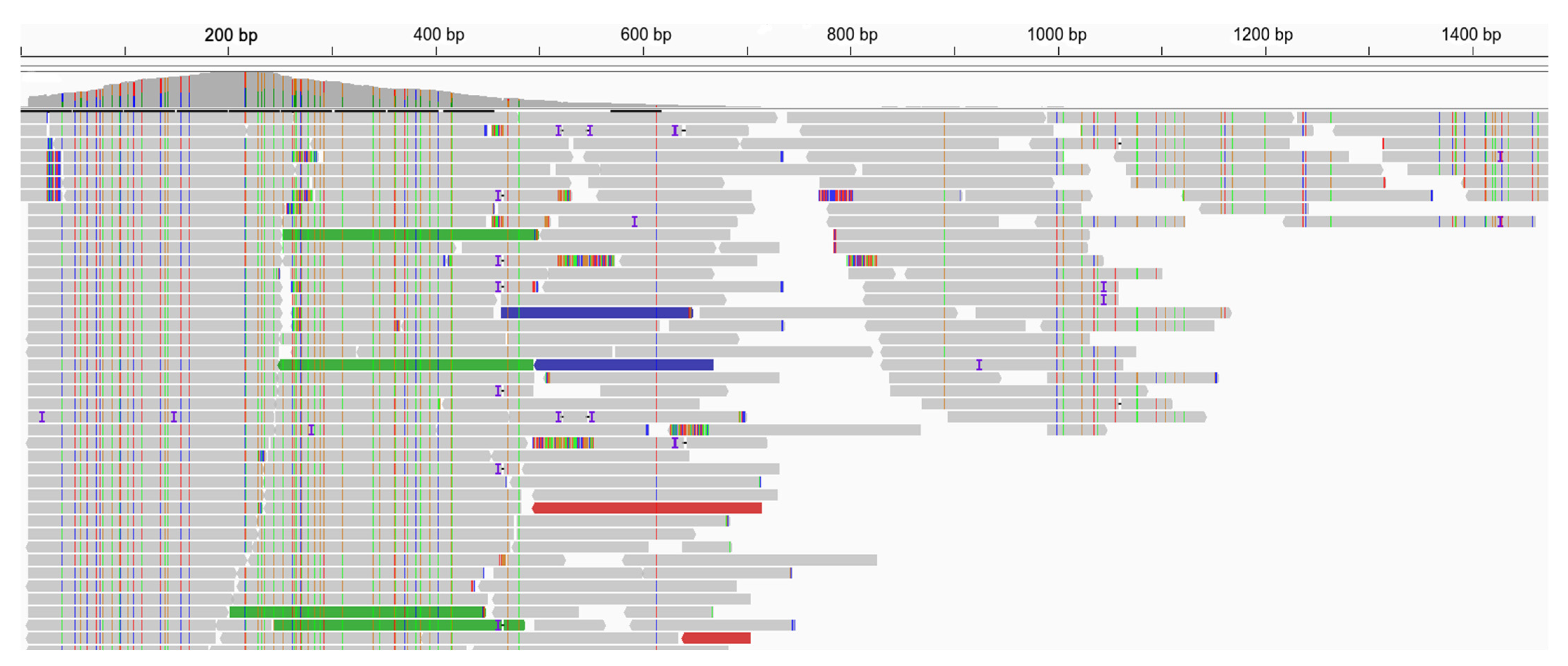
3.3. Use of the SMART Method for Amplification of Target RNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, D.; Matsvay, A.; Abramov, I.; Dedkov, V.; Shipulin, G.; Khafizov, K. Current Trends in Diagnostics of Viral Infections of Unknown Etiology. Viruses 2020, 12, 211. [Google Scholar] [CrossRef] [PubMed]
- Radford, A.D.; Chapman, D.; Dixon, L.; Chantrey, J.; Darby, A.C.; Hall, N. Application of next-generation sequencing technologies in virology. J. Gen. Virol. 2012, 93, 1853–1868. [Google Scholar] [CrossRef] [PubMed]
- Bassi, C.; Guerriero, P.; Pierantoni, M.; Callegari, E.; Sabbioni, S. Novel Virus Identification through Metagenomics: A Systematic Review. Life 2022, 12, 2048. [Google Scholar] [CrossRef]
- Koonin, E.V.; Krupovic, M.; Dolja, V.V. The global virome: How much diversity and how many independent origins? Environ. Microbiol. 2023, 25, 40–44. [Google Scholar] [CrossRef]
- Shang, J.; Sun, Y. CHEER: HierarCHical taxonomic classification for viral mEtagEnomic data via deep leaRning. Methods 2021, 189, 95–103. [Google Scholar] [CrossRef]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying viruses from metagenomic data using deep learning. Quant. Biol. 2020, 8, 64–77. [Google Scholar] [CrossRef]
- Miao, Y.; Bian, J.; Dong, G.; Dai, T. DETIRE: A hybrid deep learning model for identifying viral sequences from metagenomes. Front. Microbiol. 2023, 14, 1169791. [Google Scholar] [CrossRef]
- Bartoszewicz, J.M.; Seidel, A.; Rentzsch, R.; Renard, B.Y. DeePaC: Predicting pathogenic potential of novel DNA with reverse-complement neural networks. Bioinformatics 2020, 36, 81–89. [Google Scholar] [CrossRef]
- Kurn, N.; Chen, P.; Heath, J.D.; Kopf-Sill, A.; Stephens, K.M.; Wang, S. Novel isothermal, linear nucleic acid amplification systems for highly multiplexed applications. Clin. Chem. 2005, 51, 1973–1981. [Google Scholar] [CrossRef]
- Chrzastek, K.; Lee, D.H.; Smith, D.; Sharma, P.; Suarez, D.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 2017, 509, 159–166. [Google Scholar] [CrossRef]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; DePasse, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. Viral genome sequencing by random priming methods. BMC Genom. 2008, 9, 5. [Google Scholar] [CrossRef]
- Schmidt, W.M.; Mueller, M.W. CapSelect: A highly sensitive method for 5′ CAP-dependent enrichment of full-length cDNA in PCR-mediated analysis of mRNAs. Nucleic Acids Res. 1999, 27, e31. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.; Selleck, P.; Hooper, P.; Hyatt, A.; Gould, A.; Gleeson, L.; Westbury, H.; Hiley, L.; Selvey, L.; Rodwell, B.; et al. A morbillivirus that caused fatal disease in horses and humans. Science 1995, 268, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.B.; Bellini, W.J.; Rota, P.A.; Harcourt, B.H.; Tamin, A.; Lam, S.K.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W.-J.; et al. Nipah virus: A recently emergent deadly paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Lai, S.T.; Poon, L.L.M.; Guan, Y.; Yam, L.Y.C.; Lim, W.; Nicholls, J.; Yee, W.K.S.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Délicat, A.; Paweska, J.T.; Gonzalez, J.-P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Irving, A.T.; Ahn, M.; Goh, G.; Anderson, D.E.; Wang, L.-F. Lessons from the host defences of bats, a unique viral reservoir. Nature 2021, 589, 363–370. [Google Scholar] [CrossRef]
- Zhou, P.; Tachedjian, M.; Wynne, J.W.; Boyd, V.; Cui, J.; Smith, I.; Cowled, C.; Ng, J.H.J.; Mok, L.; Michalski, W.P.; et al. Contraction of the type I IFN locus and unusual constitutive expression of IFN-α in bats. Proc. Natl. Acad. Sci. USA 2016, 113, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz-Rivera, P.C.; Kanchwala, M.; Liang, H.; Kumar, A.; Wang, L.-F.; Xing, C.; Schoggins, J.W. The IFN Response in Bats Displays Distinctive IFN-Stimulated Gene Expression Kinetics with Atypical RNASEL Induction. J. Immunol. 2018, 200, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Leifels, M.; Khalilur Rahman, O.; Sam, I.-C.; Cheng, D.; Chua, F.J.D.; Nainani, D.; Kim, S.Y.; Ng, W.J.; Kwok, W.C.; Sirikanchana, K.; et al. The one health perspective to improve environmental surveillance of zoonotic viruses: Lessons from COVID-19 and outlook beyond. ISME Commun. 2022, 2, 107. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef]
- Juergens, K.B.; Huckabee, J.; Greninger, A.L. Two Novel Iflaviruses Discovered in Bat Samples in Washington State. Viruses 2022, 14, 994. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Y.; Machleder, E.M.; Chenchik, A.; Li, R.; Siebert, P.D. Reverse transcriptase template switching: A SMART approach for full-length cDNA library construction. Biotechniques 2001, 30, 892–897. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roev, G.V.; Borisova, N.I.; Chistyakova, N.V.; Agletdinov, M.R.; Akimkin, V.G.; Khafizov, K. Unlocking the Viral Universe: Metagenomic Analysis of Bat Samples Using Next-Generation Sequencing. Microorganisms 2023, 11, 2532. https://doi.org/10.3390/microorganisms11102532
Roev GV, Borisova NI, Chistyakova NV, Agletdinov MR, Akimkin VG, Khafizov K. Unlocking the Viral Universe: Metagenomic Analysis of Bat Samples Using Next-Generation Sequencing. Microorganisms. 2023; 11(10):2532. https://doi.org/10.3390/microorganisms11102532
Chicago/Turabian StyleRoev, German V., Nadezhda I. Borisova, Nadezhda V. Chistyakova, Matvey R. Agletdinov, Vasily G. Akimkin, and Kamil Khafizov. 2023. "Unlocking the Viral Universe: Metagenomic Analysis of Bat Samples Using Next-Generation Sequencing" Microorganisms 11, no. 10: 2532. https://doi.org/10.3390/microorganisms11102532
APA StyleRoev, G. V., Borisova, N. I., Chistyakova, N. V., Agletdinov, M. R., Akimkin, V. G., & Khafizov, K. (2023). Unlocking the Viral Universe: Metagenomic Analysis of Bat Samples Using Next-Generation Sequencing. Microorganisms, 11(10), 2532. https://doi.org/10.3390/microorganisms11102532