Abstract
A novel Gram-stain-negative, facultatively anaerobic, and non-motile bacterial strain, designated SDUM287046T, was isolated from the coastal sediments of Jingzi Port of Weihai, China. Cells of strain SDUM287046T were rod-shaped with widths of 0.4–0.5 μm and lengths of 0.7–1.4 μm and could produce flexirubin-type pigments. Optimum growth of strain SDUM287046T occurred at 33–35 °C, pH 7.0, and with 2% (w/v) NaCl. Oxidase activity was negative, but catalase activity was positive. Phylogenetic analysis based on 16S rRNA gene sequence revealed that strain SDUM287046T was most closely related to Aequorivita aquimaris D-24T (98.3%). The main cellular fatty acids were iso-C15:0, anteiso-C15:0, iso-C17:0 3–OH, and summed feature 9 (comprised of iso-C17:1 ω9c and/or C16:0 10-methyl). The sole respiratory quinone was MK-6. The polar lipids consisted of phosphatidylethanolamine (PE), one aminolipid (AL), three unidentified glycolipids (GL), and three unidentified lipids (L). The DNA G + C content was 39.3 mol%. According to the integrated results of phylogenetic, physiological, biochemical, and chemotaxonomic characteristics, we propose that strain SDUM287046T represents a novel species of the genus Aequorivita, for which the name Aequorivita aurantiaca sp. nov. is proposed. The type strain is SDUM287046T (=KCTC 92754T = MCCC 1H01418T). Comparative genomic analysis showed that the 16 Aequorivita species shared 1453 core genes and differed mainly in amino acid metabolism, cofactor metabolism, and vitamin metabolism. Biogeographic distribution analysis indicated that the marine environments were the primary habitat of Aequorivita bacteria.
1. Introduction
The marine environment, especially coastal sediment, is a very active ecosystem for material circulation and energy flow, which breeds various and abundant bacteria. However, a large number of uncultured bacteria in the ocean are difficult to culture under artificial conditions, which greatly limits the study of the ecological function and active products of marine bacteria. Therefore, it is a difficult and important task to achieve the isolation and culture of uncultured marine bacteria.
The genus Aequorivita was first proposed by Bowman and Nichols in 2002 as a member of the family Flavobacteriaceae, with A. antarctica being the type species [1], and was amended by Park et al. in 2009 [2]. At the time of writing, the genus Aequorivita consists of 18 validly published species according to the List of Prokaryotic Names with Standing in Nomenclature (LPSN, https://www.bacterio.net/species, accessed on 3 June 2023), including 7 species reclassified from the genus Vitellibacter to the genus Aequorivita [3,4,5]. Most members of the genus Aequorivita were isolated from marine environments such as seawater [1,6], marine sediment [7,8], cold seep [9], shallow water hydrothermal vent [10], seaweed [11], holothurian [12], and the intestinal tract of a squid [13], except for A. sublithincola (isolated from quartz stone subliths) [1] and A. lutea (isolated from estuarine sediment) [5]. The genus Aequorivita is widely distributed and is a potential group that can produce active compounds. In 2018, a study showed that Aequorivita sp. had antimicrobial and anthelmintic activity towards multidrug-resistant bacteria and the nematode Caenorhabditis elegans [14,15]. In addition, one strain of the genus Aequorivita was capable of producing esterase with polyethylene terephthalate (PET)-hydrolyzing activity, which suggested the potential ecological role of the genus Aequorivita in the decomposition of marine PET litter [16].
In this study, a rod-shaped, facultatively anaerobic strain was obtained from coastal sediments, and polyphasic taxonomic data suggested that the isolate can be classified as a representative of the novel species within the genus Aequorivita.
2. Materials and Methods
2.1. Bacterial Isolation and Cultivation
Strain SDUM287046T was isolated from a coastal sediment sample that was collected from Jingzi Port in Weihai, China (122°7′38.80″ E, 37°33′57.60″ N) in November 2018. The sampling depth was about 5 m underwater. The temperature of the samples was 16 °C, the salinity was 40‰, and the pH was 8.0. Approximately 10 g of sediment was added to 90 mL of sterilized seawater and shaken fully. The mixed sample solution was diluted to 10−2 with sterilized seawater using the standard 10-fold dilution technique. Then the 100 μL dilution was spread on the “sandwich agar plate” designed in our laboratory as follows: The bottom layer was marine agar 2216 (MA; Becton Dickinson, Franklin Lakes, NJ, USA), on which the bacterium Rhodovibrio salinarum DSM 9154T (Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany) was cultured as a “helper” (the middle sandwich layer), and the top layer was the modified medium (per liter: 0.1 g yeast extract, 0.5 g peptone, and 40 g agar) prepared with artificial seawater (per liter deionized water: 3.2 g MgSO4, 2.2 g MgCl2, 1.2 g CaCl2, 0.7 g KCl, 0.2 g NaHCO3, and 30 g NaCl), as described previously [17]. After the incubation at 28 °C for one week, strain SDUM287046T was isolated using plate streaking and subcultured serially on MA medium. Pure cultures were stored at –80 °C in a sterile 1% (w/v) saline solution supplemented with 20% (v/v) glycerol. The experimental strains A. aquimaris KCTC 42708T and A. antarctica DSM 14231T were obtained from the Korean Collection for Type Cultures (KCTC, Jeollabuk-do, Republic of Korea) and the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany), respectively, and were cultured under optimum conditions.
2.2. The 16S rRNA Gene Sequencing and Phylogenetic Analysis
Amplification of the 16S rRNA gene was performed using PCR technology with the primer pairs 27F and 1492R [18]. To obtain the nearly complete cloned 16S rRNA gene sequence of SDUM287046T, the clone operation was performed as described previously [19]. The purified PCR product was ligated into the pMD18-T vector (Takara Bio Inc., Dalian, China), and then the ligation product was transferred into Escherichia coli DH5a receptor cells. The 16S rRNA gene was sequenced by RuiBiotech (Qingdao, China), and alignment analysis was carried out by employing the EzBioCloud server (http://www.ezbiocloud.net/, accessed on 6 June 2023) and the NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 6 June 2023). The 16S rRNA gene phylogenetic trees were reconstructed with the neighbor-joining (NJ) [20], maximum-likelihood (ML) [21], and maximum-parsimony (ME) [22] algorithms employing the software MEGA X (version 10.2) [23]. The ML tree was reconstructed using the best-fit substitution model K2 + G + I. Bootstrap analysis was performed with 1000 replications to evaluate tree topologies.
2.3. Whole-Genome Sequencing, Genomic and Phylogenomic Analyses
Genomic DNA of strain SDUM287046T was extracted using a bacteria genomic DNA kit (Takara Bio Inc., Dalian, China) and sequenced by Novogene Bioinformatics Technology Co., Ltd. (Beijing, China) on the NovaSeq 6000 platform (Illumina, San Diego, CA, USA). The raw reads were generated by the base-calling software CASAVA (version 1.8; Illumin, San Diego, CA, USA) and filtered using the data quality control software Fastp (version 0.23.0; HaploX Biotechnology Co., Ltd., Shenzhen, China). High-quality clean data were assembled using the software SOAPdenovo2 (version r242; BGI Genomics Co., Ltd., Shenzhen, China). The genomes of other relevant strains in this study were downloaded from the NCBI prokaryotic reference genome database.
The completeness and contamination values were estimated based on the method of lineage-specific CheckM (version 1.1.6) [24], and the complete 16S rRNA genes were extracted from the genome using the algorithm ContEst16S [25]. Genome annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) based on ab initio gene prediction algorithms and homology-based methods [26]. The analysis of secondary metabolite biosynthetic gene clusters in the strain SDUM287046T genome was accomplished by the online antiSMASH server (version 7.0; https://antismash.secondarymetabolites.org/, accessed on 9 June 2023) [27].
Average nucleotide identity (ANI) and digital DNA–DNA hybridization (dDDH) values between strain SDUM287046T and experimental strains were calculated using JSpeciesWS (https://jspecies.ribohost.com/jspeciesws/, accessed on 12 June 2023) [28] and the Genome-to-Genome Distance Calculator (version 3.0; http://ggdc.dsmz.de/ggdc.php, accessed on 12 June 2023) [29], respectively. The concatenated alignment sequences of 120 ubiquitous single-copy proteins were obtained by GTDB-Tk (version 1.3.0) [30], and the phylogenetic tree was reconstructed by FastTree [31] using JTT + CAT parameters and IQ-TREE [32] using the LG + F + I + G4 model with 1000 bootstrap replicates.
2.4. Comparative Genomic Analysis and Biogeographic Distribution of the Genus Aequorivita
Genome statistics of the strains within the genus Aequorivita used in this analysis are listed in Table S1. All genomes were predicted by the Prodigal tool [33], and the metabolic pathways were analyzed in detail employing KEGG’s BlastKOALA server (https://www.kegg.jp/blastkoala/, accessed on 3 June 2023) [34]. In order to estimate the genomic diversity and identify orthologous groups among the members of the genus Aequorivita, pan-genome analysis using the bacterial pan-genome analysis (BPGA) tool was performed with default parameters (50% amino acid sequence identity) [35].
To evaluate the global distribution and habitat preference of the genus Aequorivita, the analysis pipeline Microbe Atlas Project (MAP, https://microbeatlas.org/, accessed on 30 July 2023) was used with a 96% sequence similarity threshold. The quantification of the microbial abundances in the sequenced microbial communities was based on a closed reference ribosomal RNA analysis using MAPseq [36]. The use of a common reference enabled the direct comparison of microbial taxa abundance across samples and studies using different sequencing protocols. The analyzed sequencing data included amplicon, shotgun, and metatranscriptomic sequencing.
2.5. Morphology, Physiology, and Biochemistry
The physiological and biochemical features of strain SDUM287046T were examined after incubation at 35 °C for 48 h on MA medium. Cell morphology and size were examined employing light microscopy (model E600; Nikon, Tokyo, Japan) and scanning electron microscopy (model Nova NanoSEM450; FEI, Portland, OR, USA). The Gram staining reactions were tested using crystal violet and safranin O stain solutions (Sangon Biotech Co., Ltd., Shanghai, China) according to the steps of the standard Gram reaction [37]. The presence of flexirubin-type pigments was determined using the alkaline method described previously (suspend the colonies in 20% KOH) [38,39]. The gliding motility test was carried out by preparing a cell suspension in seawater and then placing a drop on the marine 2216 medium solidified with 0.3% agar [40]. Growth range and optima of temperature were tested on MA at 0, 4, 20, 25, 28, 30, 33, 35, 37, 40, and 43 °C. Tolerance to NaCl was examined using the following medium (per liter: 1 g yeast extract and 5 g peptone), prepared with artificial seawater (per liter: 3.2 g MgSO4, 2.2 g MgCl2, 1.2 g CaCl2, 0.7 g KCl and 0.2 g NaHCO3) containing NaCl at concentrations from 0 to 10% (w/v, in 1% intervals). The pH range for growth was tested in marine broth 2216 (MB; Becton Dickinson, Franklin Lakes, NJ, USA) by adding different pH buffers at a concentration of 20 mM: MES (pH 5.5 and 6.0), PIPES (pH 6.5 and 7.0), HEPES (pH 7.5 and 8.0), Tricine (pH 8.5), and CAPSO (pH 9.0 and 9.5) (Sangon Biotech Co., Ltd., Shanghai, China). Bacterial growth monitoring was achieved with a spectrophotometer (model 900H; Perkin Elmer, Waltham, MA, USA) at 600 nm. Susceptibility to antibiotics was tested using the disc diffusion method described previously [41]. Anaerobic growth was examined after incubation for two weeks on MA in an anaerobic jar (10% H2, 10% CO2, and 80% N2).
Catalase and oxidase activities were assessed employing a 3% H2O2 solution and an oxidase test reagent (BioMérieux, Shanghai, China), respectively. Hydrolytic activity assays of starch (2%, w/v), alginate (2%, w/v), CM-cellulose (0.5%, w/v), casein (1% skimmed milk, w/v), and Tween (20, 40, 60, and 80, 1%, w/v) (Sangon Biotech Co., Ltd., Shanghai, China) were achieved following the methods described previously [42]. The API 50CH kits (BioMérieux, Shanghai, China) and Biolog GEN III MicroPlates were employed to test fermentative acid-producing activity and sole carbon source oxidation, respectively. Additional enzyme-producing activities were assessed using API ZYM and 20E kits (BioMérieux, Shanghai, China). All API and Biolog assays were performed simultaneously with the experimental strains according to the instructions, except that the NaCl concentration was adjusted to be optimal.
2.6. Chemotaxonomy
Comparative analyses of chemotaxonomic properties (fatty acids, isoprenoid quinone, and polar lipids) between strain SDUM287046T and the experimental strains were carried out using cells harvested from MB medium at the late stage of the exponential growth phase. The culture temperature of SDUM287046T was 35 °C, and the rotary shaker speed was about 200 r/min. The fatty acids were extracted by saponification, methylation, and extraction and then analyzed using a gas chromatograph (model 6890N; Agilent, Beijing, China) and the Sherlock Microbial Identification System (MIDI, version 6.1) [43]. Respiratory quinones were extracted as follows: Add 300 mg of freeze-dried pure cultures to the 40 mL mixed solution of chloroform and methanol (2:1, v/v) and stir in the dark for about 10 h; add 200 µL of acetone to dissolve after filtration and distillation operations; use silica-gel thin-layer chromatography (TLC) plates and chromatographic solution (n-hexane: ether = 85:15, v/v) to separate the quinones into different classes [44,45]. Respiratory quinones were identified using high-performance liquid chromatography (HPLC, model LC-20AT; Shimadzu, Shanghai, China). The extraction of polar lipids was achieved using a solution consisting of chloroform, methanol, and water (2.5:5:2, v/v/v), and the types of polar lipids were identified by the two-dimensional TLC method [46].
3. Results and Discussion
3.1. Phenotypic Properties
The colony of strain SDUM287046T was circular, smooth, and orange-colored. Cells of strain SDUM287046T were Gram-stain-negative and rod-shaped with widths of 0.4–0.5 μm and lengths of 0.7–1.4 μm (Figure S1). The orange-colored colonies turned orange-red in a 20% KOH solution, and the orange-red color reverted to orange after adding acid to remove the alkaline environment, which indicated that SDUM287046T could produce flexirubin-type pigments. Growth was observed at pH 6.0–9.0 (optimum, 7.0), temperatures of 16–37 °C (optimum, 33–35 °C), and in the presence of 1–5% (w/v, optimum, 2%) NaCl.
Strain SDUM287046T could hydrolyze Tweens 20, 40, and 60, but not cellulose, alginate, or agar. The activities of catalase, gelatinase, alkaline phosphatase, and acid phosphatase were positive, but the activities of urease, galactosidase, glucosidase, β-glucuronidase, cystine arylamidase, and lysine decarboxylase were negative, which was consistent with the experimental strains A. aquimaris KCTC 42708T and A. antarctica DSM 14231T. The oxidations of the sole carbon source (Biolog) were positive for dextrin, d-cellobiose, gentiobiose, d-salicin, N-acetyl-d-glucosamine, sodium lactate, gelatin, glycyl-l-proline, l-glutamic acid, methyl pyruvate, Tween-40, and sodium butyrate, but negative for d-melibiose, fucose, d-arabitol, d-aspartic acid, l-pyroglutamic acid, pectin, mucic acid, d-saccharic acid, and malic acid. Acids were produced from d-ribose, d-fructose, l-sorbose, d-turanose, d-lyxose, d-tagatose, esculin ferric citrate, and potassium 5-ketogluconate, but not from erythritol, arabinose, xylose, d-adonitol, β-methyl-d-xylopyranoside, l-rhamnose, dulcitol, d-manitol, d-sorbitol, α-methyl-d-mannopyranoside, α-methyl-d-glucopyranoside, glycogen, and d-xylitol.
Strain SDUM287046T was resistant to (μg per disc unless indicated) streptomycin (10), gentamycin (10), tobramycin (10), gentamycin (30), neomycin (30), and tetracycline (30), but sensitive to erythromycin (15), erythromycin (30), ampicillin (10), penicillin (10), chloramphenicol (30), rifampin (5), norfloxacin (30), cefotaxime sodium (30), clarithromycin (15), lincomycin (2), carbenicillin (100), and ceftriaxone (30). The differential characteristics between strain SDUM287046T and the experimental strains are summarized in Table 1.

Table 1.
Differential characteristics between strain SDUM287046T and the experimental strains.
3.2. Chemotaxonomic Characteristics
The major cellular fatty acids of strain SDUM287046T were iso-C15:0 and iso-C17:0 3–OH, which were similar to Aequorivita species [2]. Furthermore, strain SDUM287046T also contained anteiso-C15:0 and Summed Feature 9 (comprising iso-C17:1 ω9c and/or C16:0 10-methyl) (Table S1). The sole respiratory quinone was MK-6, which was consistent with that observed for the related strains. The polar lipids consisted of phosphatidylethanolamine (PE), one aminolipid (AL), three unidentified glycolipids (GL), and three unidentified lipids (L) (Figure S2). The major polar lipids of strain SDUM287046T were similar to those of experimental strains in that phosphatidylethanolamine and glycolipid were major components.
3.3. The 16S rRNA Gene Sequence and Phylogenetics
The 16S rRNA gene sequence of strain SDUM287046T (1488 bp) was aligned with the EzBioCloud database, showing that the strain had 92.6–98.3% similarity values with members of the genus Aequorivita and shared the highest with A. aquimaris KCTC 42708T (98.3%). Phylogenetic tree analysis based on 16S rRNA gene sequences showed that strain SDUM287046T was clustered into the genus Aequorivita, which could be considered to represent a novel representative of the genus Aequorivita (Figure 1). The similar topologies of strain SDUM287046T and related species were also obtained in the phylogenetic trees reconstructed with the ML and ME algorithms.
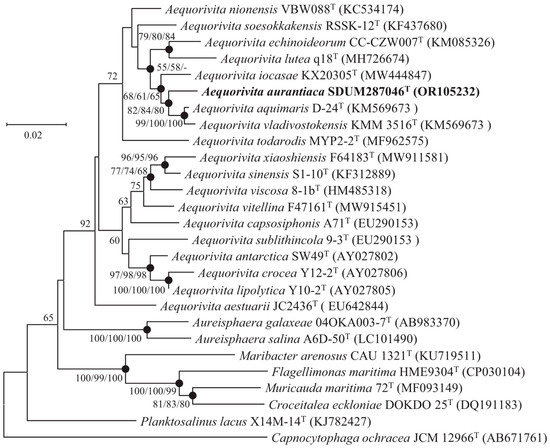
Figure 1.
Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences of strain SDUM287046T and other closely related species. Filled circles indicate branches that were recovered with neighbor-joining, maximum-likelihood, and minimum-evolution methods. Bootstrap values above 50% (1000 replicates) are shown at branch nodes (NJ/ML/ME). Capnocytophaga ochracea JCM 12966T was used as the outgroup. Bar: 0.02 substitutions per nucleotide position.
3.4. Genomic Features and Phylogenomics
The draft genome of strain SDUM287046T had a total length of 3,093,921 bp with 53 scaffolds after assembly. The DNA G + C content was estimated to be 39.3 mol%. Genome completeness and contamination values were 98.9% and 0.5%, respectively. One 16S rRNA gene sequence (1524 bp) was detected from the genome, which has 100% similarity with the cloned 16S rRNA gene sequence obtained from amplification. According to the results of PGAP, the genome of strain SDUM287046T contained 2878 genes, including 2825 protein-coding genes, 11 pseudogenes, and 42 RNA genes (3 rRNA, 35 tRNA, and 4 ncRNA). According to the results of antiSMASH, three secondary metabolite biosynthetic gene clusters encoding the types of aryl polyene, resorcinol, Type III polyketide synthase (PKS), and terpene were predicted in the genome of strain SDUM287046T. Among them, the gene cluster encoding aryl polyene and resorcinol types had the highest similarity value (75%) with the known flexirubin biosynthetic gene cluster from Flavobacterium johnsoniae UW101. The gene cluster encoding terpene type shared a 28% similarity value with the known carotenoid biosynthetic gene cluster from Algoriphagus sp. KK10202C.
The ANI and dDDH values between strain SDUM287046T and A. aquimaris D-24T (A. antarctica DSM 14231T) were 77.6% (78.3%) and 20.8% (21.5%), respectively, which were lower than the species delineation thresholds of 95–96% for ANI and 70% for dDDH [47,48]. The protein phylogenetic tree, showing the evolutionary relationships of strain SDUM287046T and some related type strains, indicated the strain was affiliated with the genus Aequorivita (Figure 2), which was consistent with the result of 16S rRNA gene phylogenetic analysis.
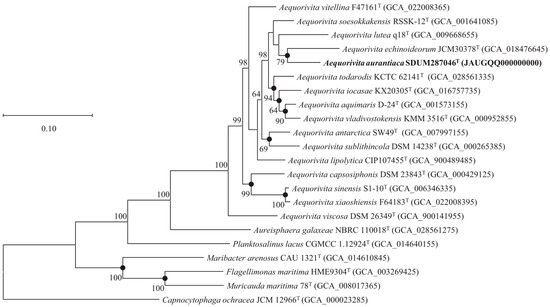
Figure 2.
The FastTree is based on 120 ubiquitous single-copy proteins. Bootstrap values above 50% (1000 replicates) are shown at branch nodes. Filled circles indicate that the same topology is also obtained using the IQ-TREE algorithm. Capnocytophaga ochracea JCM 12966T was used as the outgroup. Bar: 0.10 substitutions per nucleotide position.
3.5. Comparative Genomic Analysis of the Genus Aequorivita
The size of all Aequorivita genomes, including strain SDUM287046T, ranged from 2,929,928 bp to 4,042,904 bp, and the GC contents ranged from 34.5% to 42.8% (Table S2). As presented in Figure 3, the pan-genome analysis based on orthologous groups of proteins revealed that 1453 core genes were shared by the 16 Aequorivita strains, which was about half of each genome. The percentage of accessory genes in each Aequorivita genome ranged from 28.8% to 44.9% and unique genes from 5.9% to 24.9%. Moreover, the analysis of KEGG distribution showed that the core genes were more involved in the metabolisms supporting basic life activities, such as nucleotide metabolism and translation, while unique genes were more distributed in carbohydrate metabolism, cell motility, cellular community, and membrane transport pathways (Figure S3), which might give the genus Aequorivita species metabolic diversity and flexibility [19].
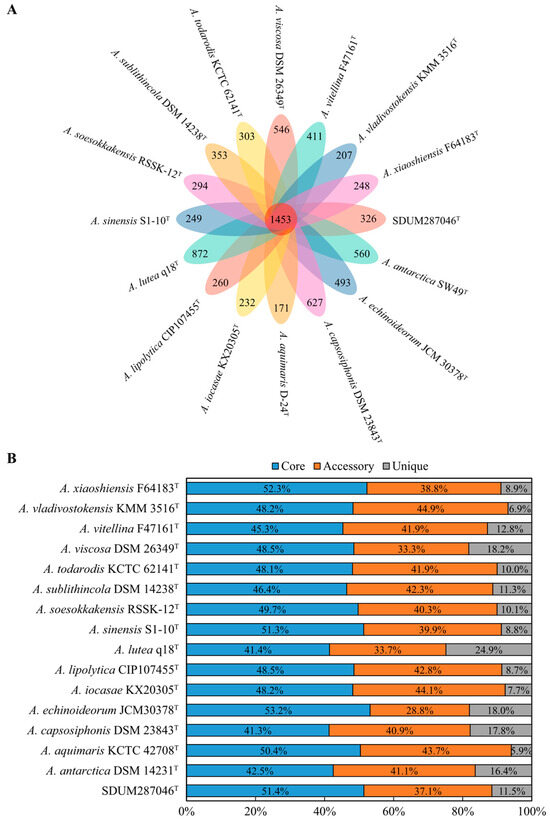
Figure 3.
Pan-genome analysis of the genus Aequorivita (16 genomes). (A) Venn diagram displaying the numbers of core gene families and unique genes for each Aequorivita strain. (B) Percentage of core, accessory, and unique genes in each genome.
According to the KEGG annotation analysis (Figure 4), the TCA cycle pathway (M00009) and F-type ATPase (M00157) were complete in all Aequorivita genomes, while the glycolysis pathway (M00001) was not. Incomplete glycolysis pathways kept Aequorivita strains from fermenting with glucose as the sole carbon source in anoxic environments, which was consistent with the results of API 50CH kits. Considering that pyruvate deficiency due to glycolysis pathway incompleteness leads to a lack of acetyl-CoA from the pyruvate oxidation pathway, fatty acid oxidation and amino acid metabolism may be the important sources of acetyl-CoA for the Aequorivita strains. The phosphatidylethanolamine (PE) biosynthesis pathway (M00093) was annotated in all Aequorivita strains, which accorded with the result of the phenotypic experiment (Figure S2). The Aequorivita species were relatively conservative in energy metabolism, lipid metabolism, and nucleotide metabolism and differed mainly in amino acid metabolism, cofactor metabolism, and vitamin metabolism. For proline metabolism pathways, all 16 members of the genus Aequorivita had a complete proline degradation pathway (M00970), but only A. antarctica DSM 14231T, A. capsosiphonis DSM 23843T, A. lipolytica CIP107455T, A. sinensis S1-10T, A. echinoideorum JCM30378T, A. viscosa DSM 26349T, A. vitellina F47161T, and A. xiaoshiensis F64183T had a complete proline biosynthesis pathway (M00015). Proline is an effective compatible solute that can resist the adverse effects of hypertonic and low-temperature environments on cells [49,50], and proline is involved in regulating the balance of reactive oxygen species, providing oxidative stress protection to cells [51]. However, excess proline was detrimental to cell growth, and intracellular proline must be present at appropriate levels [52]. Differences in proline metabolism may provide opportunities for the genus Aequorivita to adapt to various environmental conditions. The complete histidine degradation pathway (M00045) was found in all genomes, but the intact histidine biosynthesis pathway (M00026) was not annotated in A. aquimaris D-24T, A. sinensis S1-10T, A. echinoideorum JCM30378T, A. viscosa DSM 26349T, and A. xiaoshiensis F64183T.

Figure 4.
The metabolic module integrity of Aequorivita strains. The solid circles and hollow circles indicate that the metabolic pathways were complete and incomplete, respectively, as shown in the legend.
3.6. Biogeographic Distribution of the Genus Aequorivita
The global distribution of the genus Aequorivita was analyzed using the MAP database and pipeline, and the representative sequence was found in 19,335 samples from 3429 projects (details of the samples are summarized in Sheet S1). The results of biogeographic distribution analysis showed that members of the genus Aequorivita were widely distributed, including in aquatic environments, soil environments, animal environments, and plant environments. The Aequorivita bacteria were detected in 6923 aquatic samples (35.8%), but only 376 samples related plants (1.94%), and of the known aquatic environments, marine environments were the primary habitat (Figure 5A), which corresponded to the situation that most isolates were isolated from marine environments. The quantitative analysis of the database sequencing reads mapping to the representative OTU sequence showed the known habitat with the highest number of reads related to the genus Aequorivita was the marine environment (13.2%), followed by bird gut (9.0%) (Figure 5B).
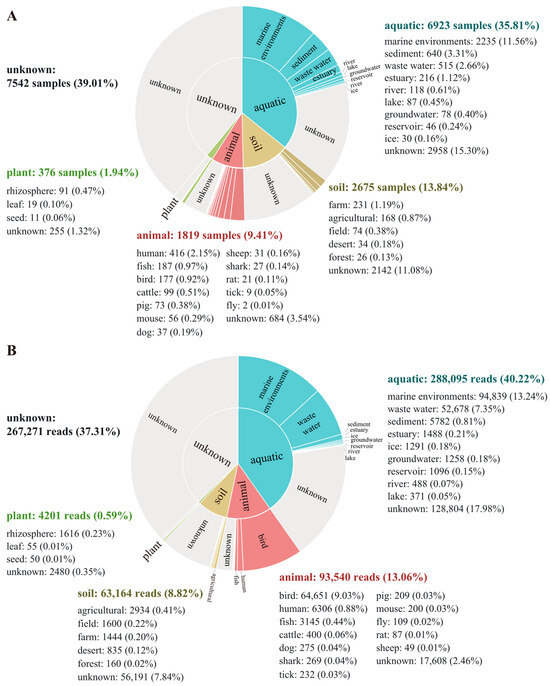
Figure 5.
Biogeographic distribution analysis of the genus Aequorivita based on the Microbe Atlas Project (MAP) database and pipeline. (A) Number of samples containing the representative OTU sequence, per habitat and sub-habitat. (B) Number of sequencing reads mapping to the representative OTU sequence, per habitat and sub-habitat.
3.7. Description of Aequorivita aurantiaca sp. nov.
Aequorivita aurantiaca (au.ran’ti.a’ca. N.L. fem. adj. aurantiaca, orange-colored, pertaining to the orange color of colonies).
Cells are rod-shaped and facultatively anaerobic with a positive Voges–Proskauer reaction. Nitrate cannot be reduced to nitrite, but flexirubin-type pigments are produced. The activities of arginine dihydrolase, tryptophan deaminase, gelatinase, esterase (C4), esterase lipase (C8), trypsin, and ɑ-chymotrypsin are positive, while those of cystine arylamidase, β-glucuronidase, galactosidase, glucosidase, N-acetyl-β-glucosaminidase, ɑ-mannosidase, and ɑ-fucosidase are negative. The major cellular fatty acids are iso-C15:0, anteiso-C15:0, iso-C17:0 3–OH, and summed feature 9 (comprised iso-C17:1 ω9c and/or C16:0 10-methyl). The sole respiratory quinone is MK-6. The polar lipids consist of phosphatidylethanolamine (PE), one aminolipid (AL), three unidentified glycolipids (GL), and three unidentified lipids (L). The DNA G + C content of the type strain is 39.3 mol%. The type strain is SDUM287046T (=KCTC 92754T = MCCC 1H01418T), which was isolated from coastal sediment samples of Jingzi Port in Weihai, China.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms11102518/s1, Figure S1: The distribution of core genes, accessory genes, and unique genes to different metabolic pathways in the genus Aequorivita. Figure S2: Scanning electron micrograph of cells of strain SDUM287046T. Bar: 5 μm. Figure S3: Two-dimensional TLC plate image of the total polar lipids of strain SDUM287046T (a), A. antarctica DSM 14231T (b), and A. aquimaris KCTC 42708T (c). PE, phosphatidylethanolamine; PGL, phosphoglycolipid; AL, unidentified aminolipid; GL, unidentified glycolipid; L, unidentified lipid. Table S1: Genomic dataset of the strains analyzed in the pan-genome analysis. Table S2: Cellular fatty acid composition (%) of the strain SDUM287046T and experimental strains. Sheet S1: Detailed information of the 19,335 samples (from 3429 projects) for biogeographic distribution analysis.
Author Contributions
J.-C.L., Y.-Q.Y. and X.-Y.T. performed experimental operation. Y.-Q.Y. performed data processing and analysis and finished the manuscript. M.-Q.Y. and Z.-J.D. offered experiment guidance and critical revision of manuscripts. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (32200003, 32070002), the Natural Science Foundation of Shandong Province (ZR2022QC106), the Guangdong Basic and Applied Basic Research Foundation (2022A1515110773), and the National Science and Technology Fundamental Resources Investigation Program of China (2019FY100700, 2022FY101100).
Data Availability Statement
The GenBank accession number of Aequorivita aurantiaca SDUM287046T for the 16S rRNA gene sequence is OR105232, for the whole-genome assembly is JAUGQQ000000000.
Acknowledgments
The scanning electron microscopy (model Nova NanoSEM450) was supported by the Physical-Chemical Materials Analytical and Testing Center of Shandong University at Weihai.
Conflicts of Interest
The authors declare that there are no conflict of interest.
References
- Bowman, J.P.; Nichols, D.S. Aequorivita gen. nov., a member of the family Flavobacteriaceae isolated from terrestrial and marine Antarctic habitats. Int. J. Syst. Evol. Microbiol. 2002, 52, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C.; Baik, K.S.; Kim, M.S.; Kim, S.S.; Kim, S.R.; Oh, M.-J.; Kim, D.; Bang, B.-H.; Seong, C.N. Aequorivita capsosiphonis sp. nov., isolated from the green alga Capsosiphon fulvescens, and emended description of the genus Aequorivita. Int. J. Syst. Evol. Microbiol. 2009, 59, 724–728. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hahnke, R.L.; Meier-Kolthoff, J.P.; García-López, M.; Mukherjee, S.; Huntemann, M.; Ivanova, N.N.; Woyke, T.; Kyrpides, N.C.; Klenk, H.-P.; Göker, M. Genome-Based Taxonomic Classification of Bacteroidetes. Front. Microbiol. 2016, 7, 2003. [Google Scholar] [CrossRef]
- García-López, M.; Meier-Kolthoff, J.P.; Tindall, B.J.; Gronow, S.; Woyke, T.; Kyrpides, N.C.; Hahnke, R.L.; Göker, M. Analysis of 1000 Type-Strain Genomes Improves Taxonomic Classification of Bacteroidetes. Front. Microbiol. 2019, 10, 2083. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, H.; Sun, C.; Hu, Z.; Wang, H. Aequorivita lutea sp. nov., a novel bacterium isolated from the estuarine sediment of the Pearl River in China, and transfer of Vitellibacter todarodis and Vitellibacter aquimaris to the genus Aequorivita as Aequorivita todarodis comb. nov. and Aequorivita aquimaris comb. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 3117–3122. [Google Scholar] [CrossRef]
- Thevarajoo, S.; Selvaratnam, C.; Goh, K.M.; Hong, K.W.; Chan, X.Y.; Chan, K.G.; Chong, C.S. Vitellibacter aquimaris sp. nov., a marine bacterium isolated from seawater. Int. J. Syst. Evol. Microbiol. 2016, 66, 3662–3668. [Google Scholar] [CrossRef]
- Wang, Q.; Cai, S.D.; Liu, J.; Zhang, D.C. Aequorivita sinensis sp. nov., isolated from sediment of the East China Sea, and reclassification of Vitellibacter todarodis as Aequorivita todarodis comb. nov. and Vitellibacter aquimaris as Aequorivita aquimaris comb. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 3323–3327. [Google Scholar] [CrossRef]
- Wang, Y.W.; Zhang, J.; Wang, S.X.; Du, Z.J.; Mu, D.S. Aequorivita vitellina sp. nov. and Aequorivita xiaoshiensis sp. nov., isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 2023, 73, 005801. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, H.; Cao, L.; Chen, H.; Zhong, Z.; Wang, M.; Li, C. Aequorivita iocasae sp. nov., a halophilic bacterium isolated from sediment collected at a cold seep field in the South China Sea. Int. J. Syst. Evol. Microbiol. 2022, 72, 005199. [Google Scholar] [CrossRef]
- Rajasabapathy, R.; Mohandass, C.; Yoon, J.H.; Dastager, S.G.; Liu, Q.; Khieu, T.N.; Son, C.K.; Li, W.J.; Colaço, A. Vitellibacter nionensis sp. nov., isolated from a shallow water hydrothermal vent. Int. J. Syst. Evol. Microbiol. 2015, 65, 692–697. [Google Scholar] [CrossRef]
- Liu, J.J.; Zhang, X.Q.; Pan, J.; Sun, C.; Zhang, Y.; Li, C.Q.; Zhu, X.F.; Wu, M. Aequorivita viscosa sp. nov., isolated from an intertidal zone, and emended descriptions of Aequorivita antarctica and Aequorivita capsosiphonis. Int. J. Syst. Evol. Microbiol. 2013, 63, 3192–3196. [Google Scholar] [CrossRef] [PubMed]
- Nedashkovskaya, O.I.; Suzuki, M.; Vysotskii, M.V.; Mikhailov, V.V. Vitellibacter vladivostokensis gen. nov., sp. nov., a new member of the phylum Cytophaga-Flavobacterium-Bacteroides. Int. J. Syst. Evol. Microbiol. 2003, 53, 1281–1286. [Google Scholar] [CrossRef]
- Kim, H.C.; Kim, Y.O.; Park, S.; Nam, B.H.; Kim, D.G.; Park, J.M.; Yoon, J.H. Vitellibacter todarodis sp. nov., isolated from intestinal tract of a squid (Todarodes pacificus). Int. J. Syst. Evol. Microbiol. 2018, 68, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Chianese, G.; Esposito, F.P.; Parrot, D.; Ingham, C.; De Pascale, D.; Tasdemir, D. Linear Aminolipids with Moderate Antimicrobial Activity from the Antarctic Gram-Negative Bacterium Aequorivita sp. Mar. Drugs 2018, 16, 187. [Google Scholar] [CrossRef] [PubMed]
- Palma Esposito, F.; Ingham, C.J.; Hurtado-Ortiz, R. Isolation by Miniaturized Culture Chip of an Antarctic bacterium Aequorivita sp. with antimicrobial and anthelmintic activity. Biotechnol. Rep. 2018, 20, e281. [Google Scholar] [CrossRef]
- Zhang, H.; Perez-Garcia, P.; Dierkes, R.F.; Applegate, V.; Schumacher, J.; Chibani, C.M.; Sternagel, S.; Preuss, L.; Weigert, S.; Schmeisser, C.; et al. The Bacteroidetes Aequorivita sp. and Kaistella jeonii Produce Promiscuous Esterases with PET-Hydrolyzing Activity. Front. Microbiol. 2021, 12, 803896. [Google Scholar] [CrossRef]
- Lian, F.B.; Jiang, S.; Zhu, K.L.; Shang, D.D.; Zhang, J.; Du, Z.J. Salegentibacter maritimus sp. nov., isolated from marine coastal sediment. Syst. Appl. Microbiol. 2021, 44, 126209. [Google Scholar] [CrossRef]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; Wiley: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Ye, Y.Q.; Hao, Z.P.; Yue, Y.Y.; Ma, L.; Ye, M.Q.; Du, Z.J. Characterization of Kordiimonas marina sp. nov. and Kordiimonas laminariae sp. nov. and comparative genomic analysis of the genus Kordiimonas, a marine-adapted taxon. Front. Mar. Sci. 2022, 9, 919253. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Kumar, S. A stepwise algorithm for finding minimum evolution trees. Mol. Biol. Evol. 1996, 13, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Lee, I.; Chalita, M.; Ha, S.M.; Na, S.I.; Yoon, S.H.; Chun, J. ContEst16S: An algorithm that identifies contaminated prokaryotic genomes using 16S RNA gene sequences. Int. J. Syst. Evol. Microbiol. 2017, 67, 2053–2057. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; De Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA—An ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef]
- Matias Rodrigues, J.F.; Schmidt, T.S.; Tackmann, J.; von Mering, C. MAPseq: Highly efficient k-mer search with confidence estimates, for rRNA sequence analysis. Bioinformatics 2017, 33, 3808–3810. [Google Scholar] [CrossRef]
- Smibert, R.M.; Krieg, N.R. Phenotypic characterization. In Methods for General and Molecular Bacteriology; Gerhardt, P., Murray, R., Wood, W., Krieg, N.R., Eds.; American Society for Microbiology: Washington, DC, USA, 1994; pp. 607–654. [Google Scholar]
- Fautz, E.; Reichenbach, H. A simple test for flexirubin-type pigments. FEMS Microbiol. Lett. 1980, 8, 87–91. [Google Scholar] [CrossRef]
- Bernardet, J.; Bowman, J. The genus Flavobacterium. In The Prokaryotes, a Handbook on the Biology of Bacteria, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; Volume 7, pp. 481–531. [Google Scholar]
- Bowman, J.P. Description of Cellulophaga algicola sp. nov., isolated from the surfaces of Antarctic algae, and reclassification of Cytophaga uliginosa (ZoBell and Upham 1944) Reichenbach 1989 as Cellulophaga uliginosa comb. nov. Int. J. Syst. Evol. Microbiol. 2000, 50, 1861–1868. [Google Scholar] [CrossRef]
- Jorgensen, J.H.; Turnidge, J.D. Susceptibility test methods. In Manual of Clinical Microbiology, 11th ed.; Jorgensen, J.H., Carroll, K.C., Funke, G., Pfaller, M.A., Landry, M.L., Richter, S.S., Warnock, D.W., Eds.; Wiley: New York, NY, USA, 2015; pp. 1253–1273. ISBN 9781683672807. [Google Scholar]
- Dong, X.Z.; Cai, M.Y. Determination of biochemical characteristics. In Manual for the Systematic Identification of General Bacteria, 14th ed.; Dong, X.Z., Cai, M.Y., Eds.; Science Press: Beijing, China, 2001; pp. 370–398. [Google Scholar]
- Sasser, M. Identification of Bacteria by Gas Chromatography of Cellular Fatty Acids; MIDI Inc.: Newark, DE, USA, 1990. [Google Scholar]
- Minnikin, D.E.; O’donnell, A.G.; Goodfellow, M.; Alderson, G.; Athalye, M.; Schaal, A.; Parlett, J.H. An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Meth. 1984, 2, 233–241. [Google Scholar] [CrossRef]
- Hiraishi, A.; Ueda, Y.; Ishihara, J.; Mori, T. Comparative lipoquinone analysis of influent sewage and activated sludge by high-performance liquid chromatography and photodiode array detection. J. Gen. Appl. Microbiol. 1996, 42, 457–469. [Google Scholar] [CrossRef]
- Komagata, K.; Suzuki, K. Lipid and cell-wall analysis in bacterial systematics. Method Microbiol. 1987, 19, 161–207. [Google Scholar]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Hoffmann, T.; Von Blohn, C.; Stanek, A.; Moses, S.; Barzantny, H.; Bremer, E. Synthesis, release, and recapture of compatible solute proline by osmotically stressed Bacillus subtilis cells. Appl. Environ. Microb. 2012, 78, 5753–5762. [Google Scholar] [CrossRef]
- Ting, L.; Williams, T.J.; Cowley, M.J.; Lauro, F.M.; Guilhaus, M.; Raftery, M.J.; Cavicchioli, R. Cold adaptation in the marine bacterium, Sphingopyxis alaskensis, assessed using quantitative proteomics. Environ. Microbiol. 2010, 12, 2658–2676. [Google Scholar] [CrossRef]
- Kaul, S.; Sharma, S.S.; Mehta, I.K. Free radical scavenging potential of L-proline: Evidence from in vitro assays. Amino Acids 2008, 34, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Takagi, H. Role of the yeast acetyltransferase Mpr1 in oxidative stress: Regulation of oxygen reactive species caused by a toxic proline catabolism intermediate. Proc. Natl. Acad. Sci. USA 2004, 101, 12616–12621. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).