
Uncovering Microbiome Adaptations in a Full-Scale Biogas Plant: Insights from MAG-Centric Metagenomics and Metaproteomics

 , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
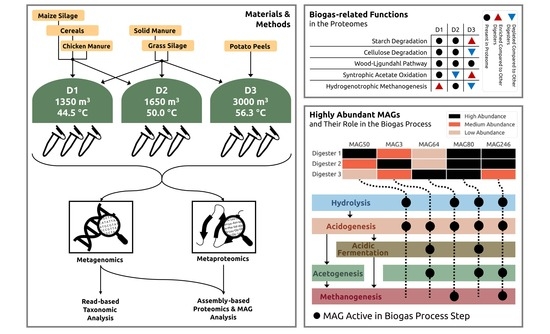
2.1. Sampling at the Full-Scale Biogas Plant and Metadata Compilation
2.2. DNA Isolation, Metagenome Library Preparation and Sequencing
2.3. Bioinformatic Processing, Single-Read Analyses and Assembly/Binning of the Metagenomic Datasets
2.4. Generation, Processing and Analyses of the Metaproteomic Datasets
2.5. Metagenomically Assembled Genome (MAG)-Centric Metagenome and Metaproteome Analysis
3. Results and Discussion
3.1. The Full-Scale Biogas Plant 35 Consists of Three Parallel Lines Differing in the General Process Operation, Especially Feedstocks and Process Temperature
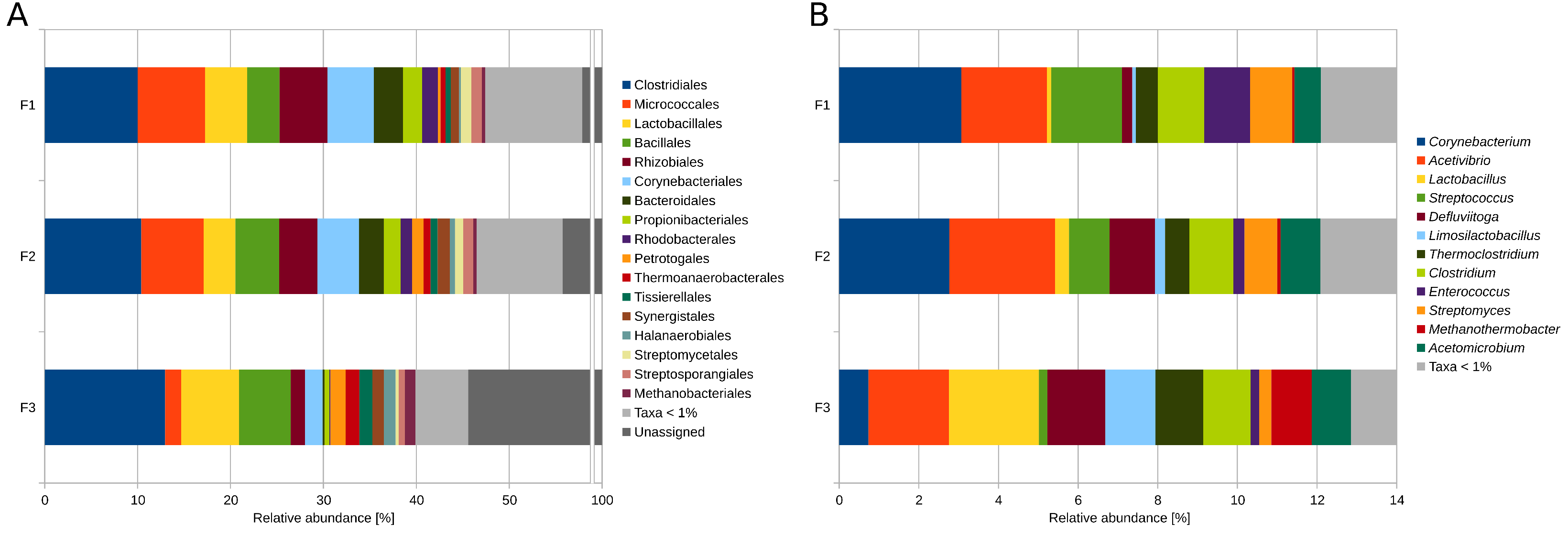
3.2. Taxonomic Profiling and Functional Potential Based on Metagenome Single-Read Analyses
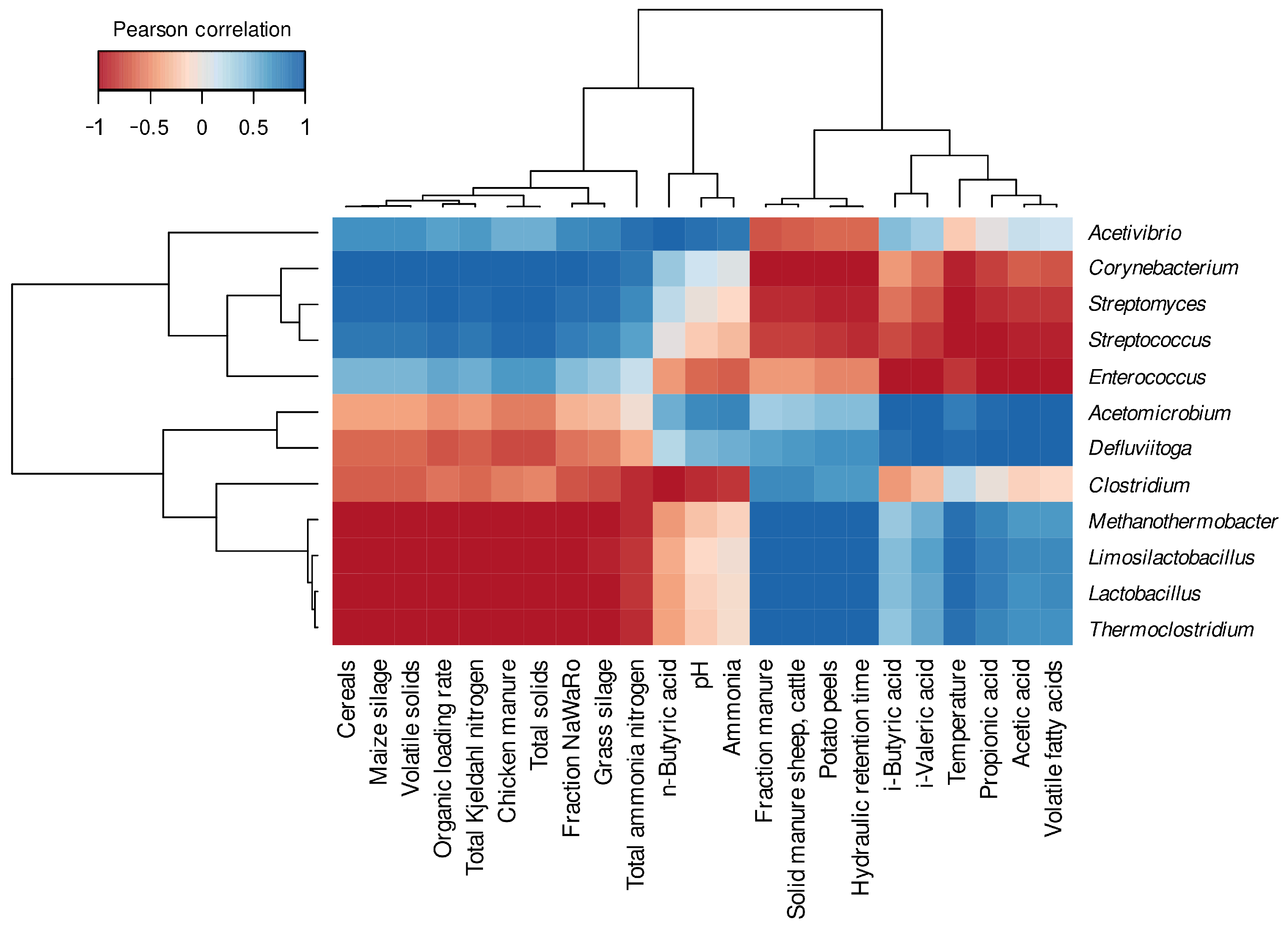
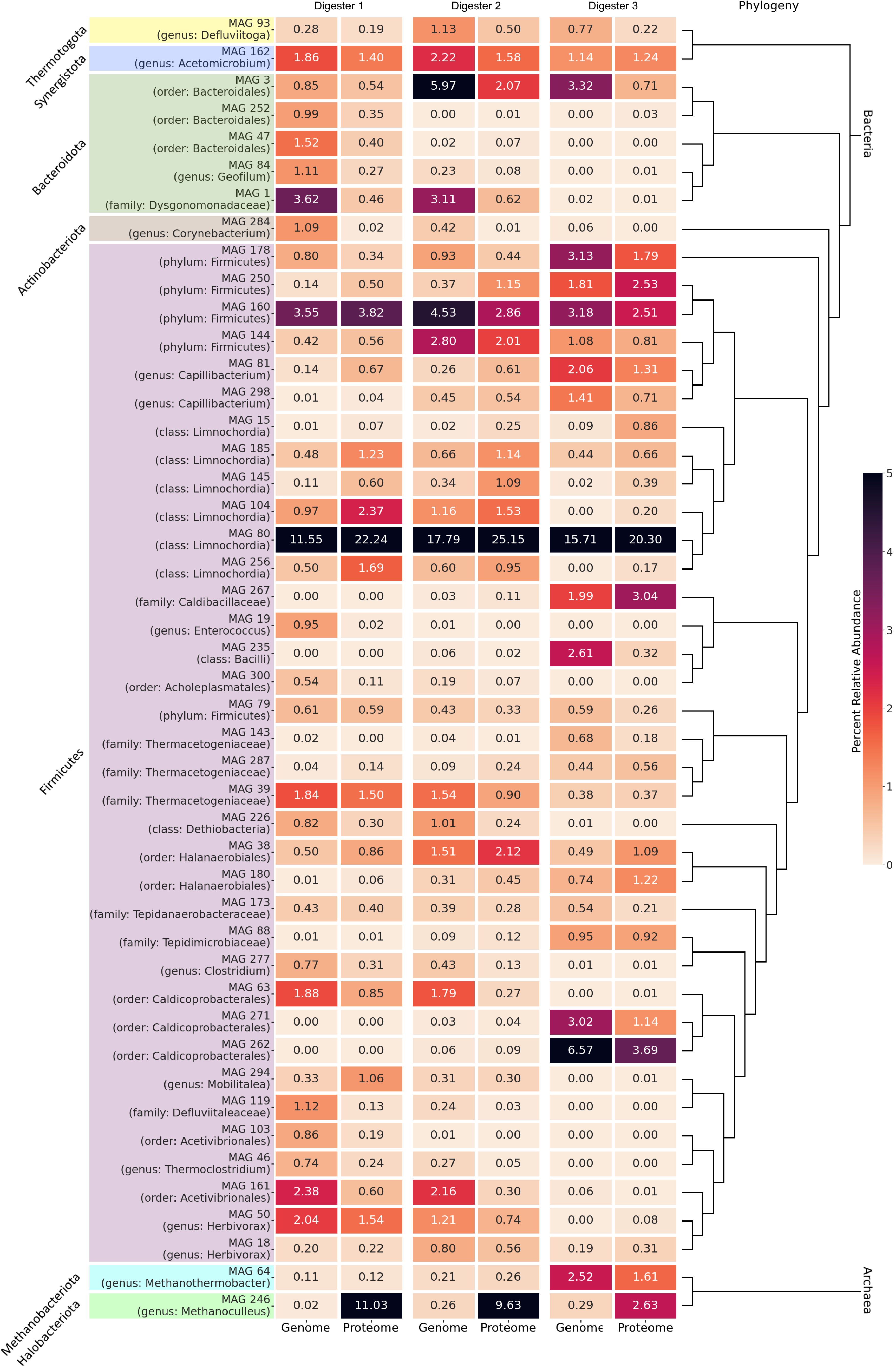
3.2.1. Differentially Abundant and Evenly Distributed Taxa Residing in the Three Digesters as Revealed by Taxonomic Profiling Based on Metagenome Single-Read Analyses
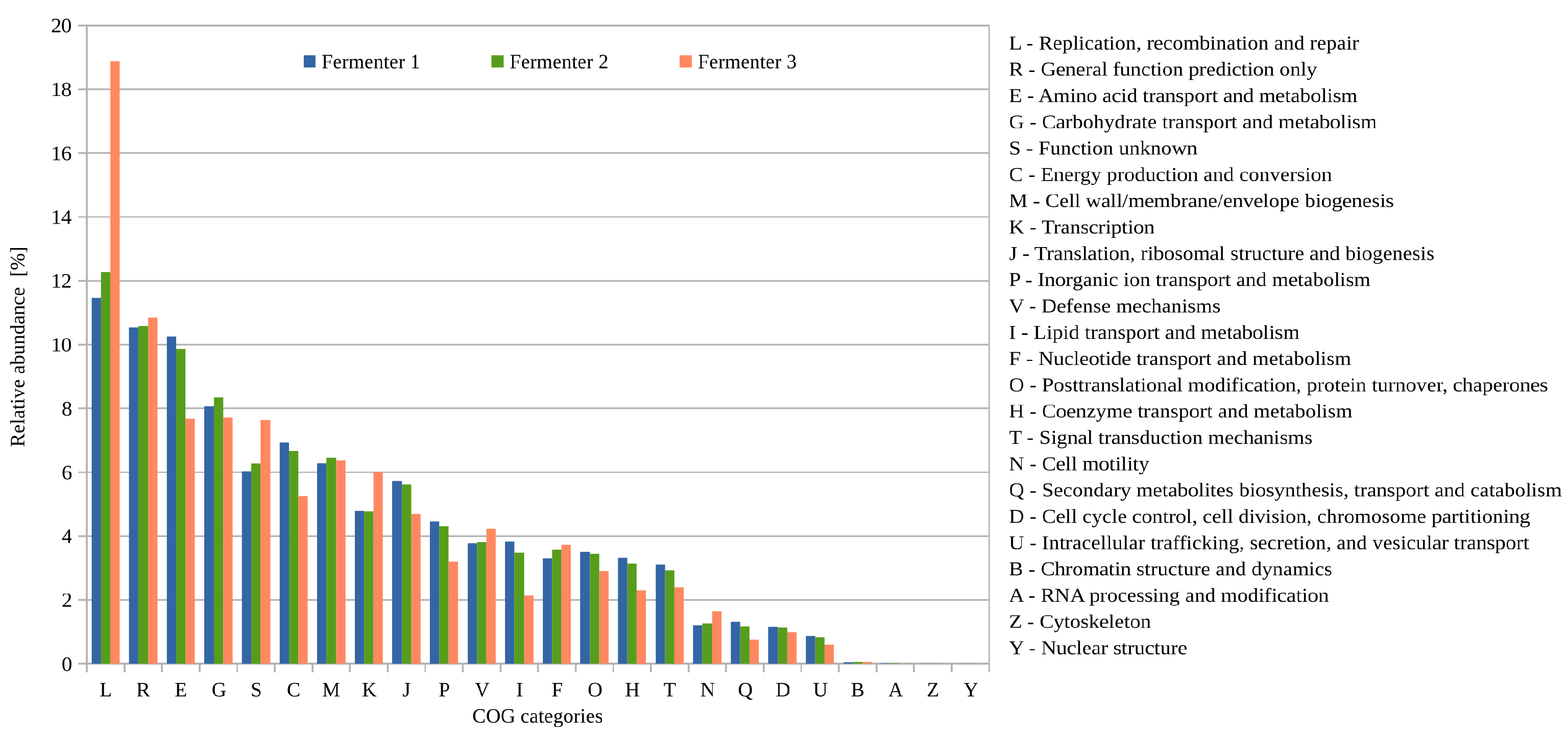
3.2.2. Functional Potential (COG) of the Biogas Microbiomes Residing in the Three Digesters of Biogas Plant 35 Based on Single-Read Analyses
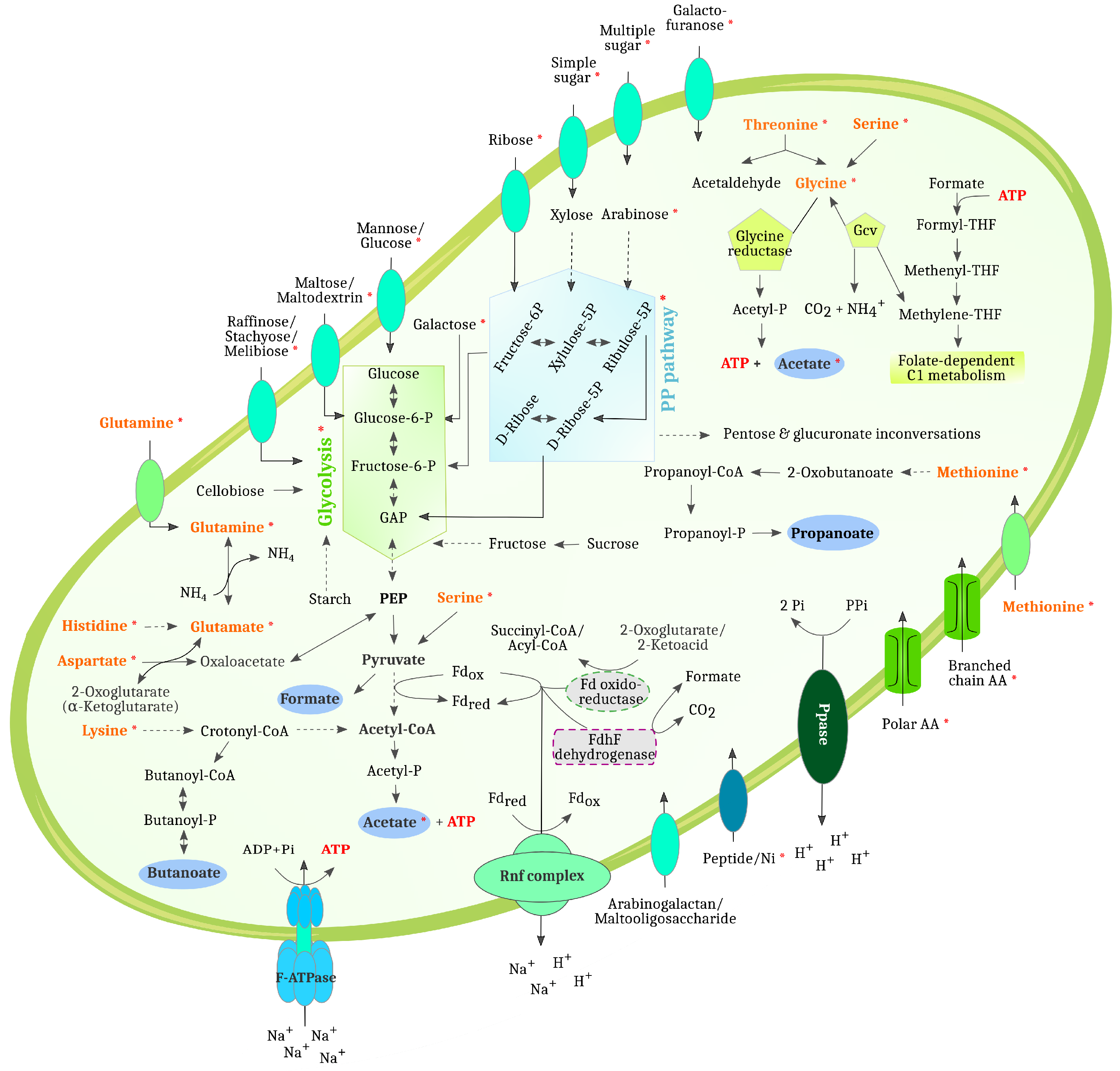
3.3. Biogas Process-Related Functional Potential and Expressed Functions Resulting from Metagenome and Metaproteome Analyses Indicate Microbial Community Adaptations to the Process Conditions in the Three Digesters
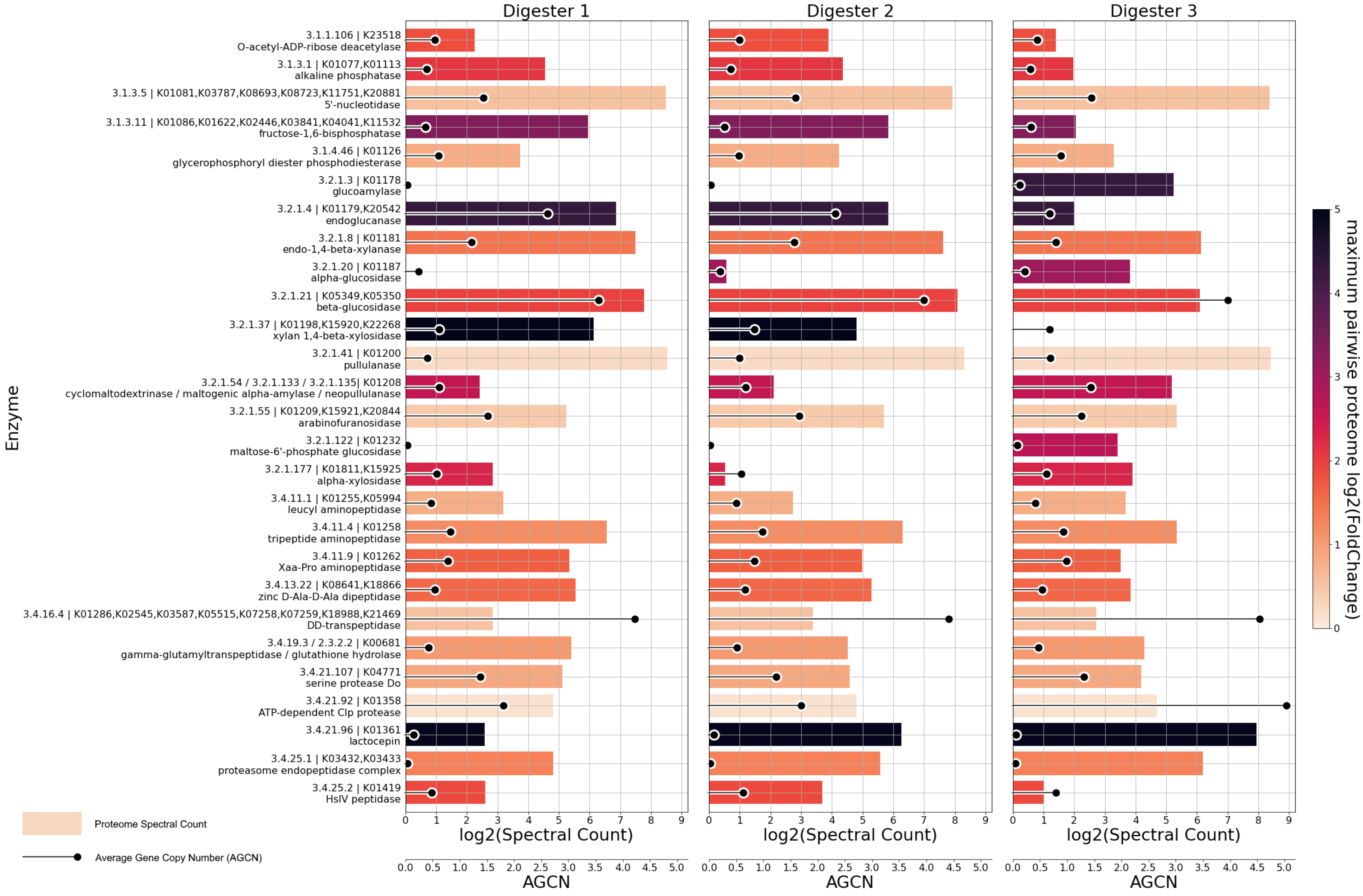
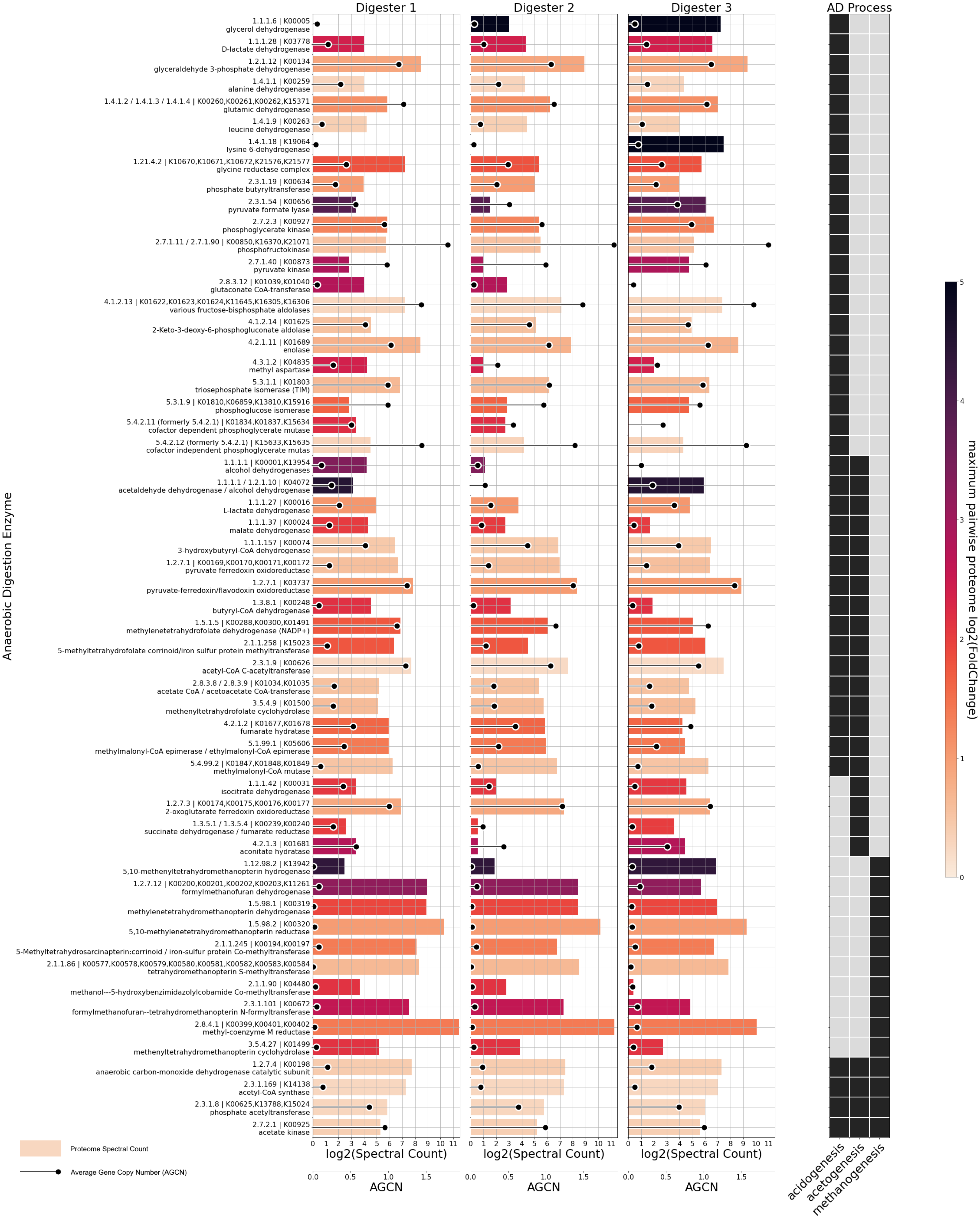
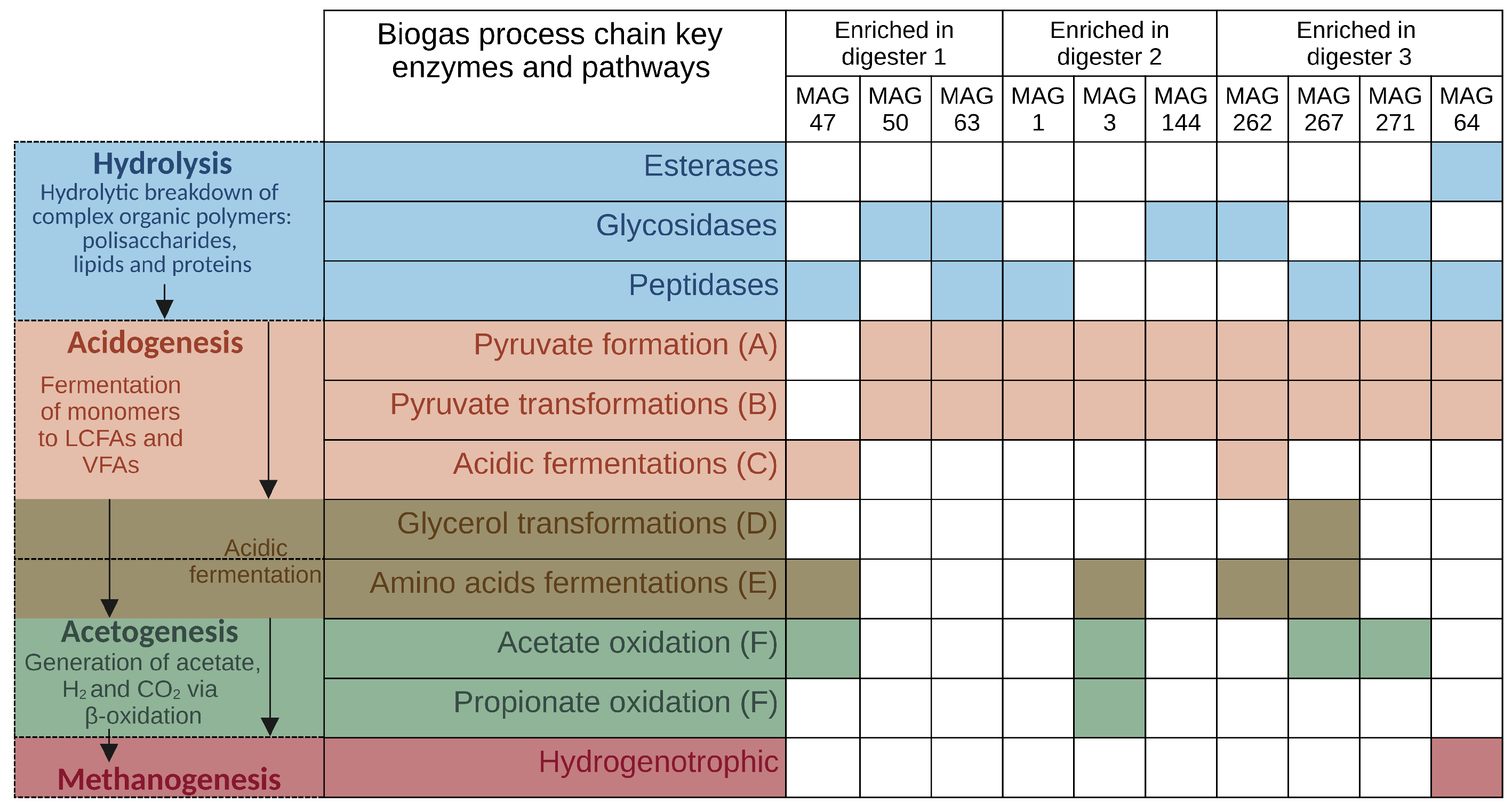
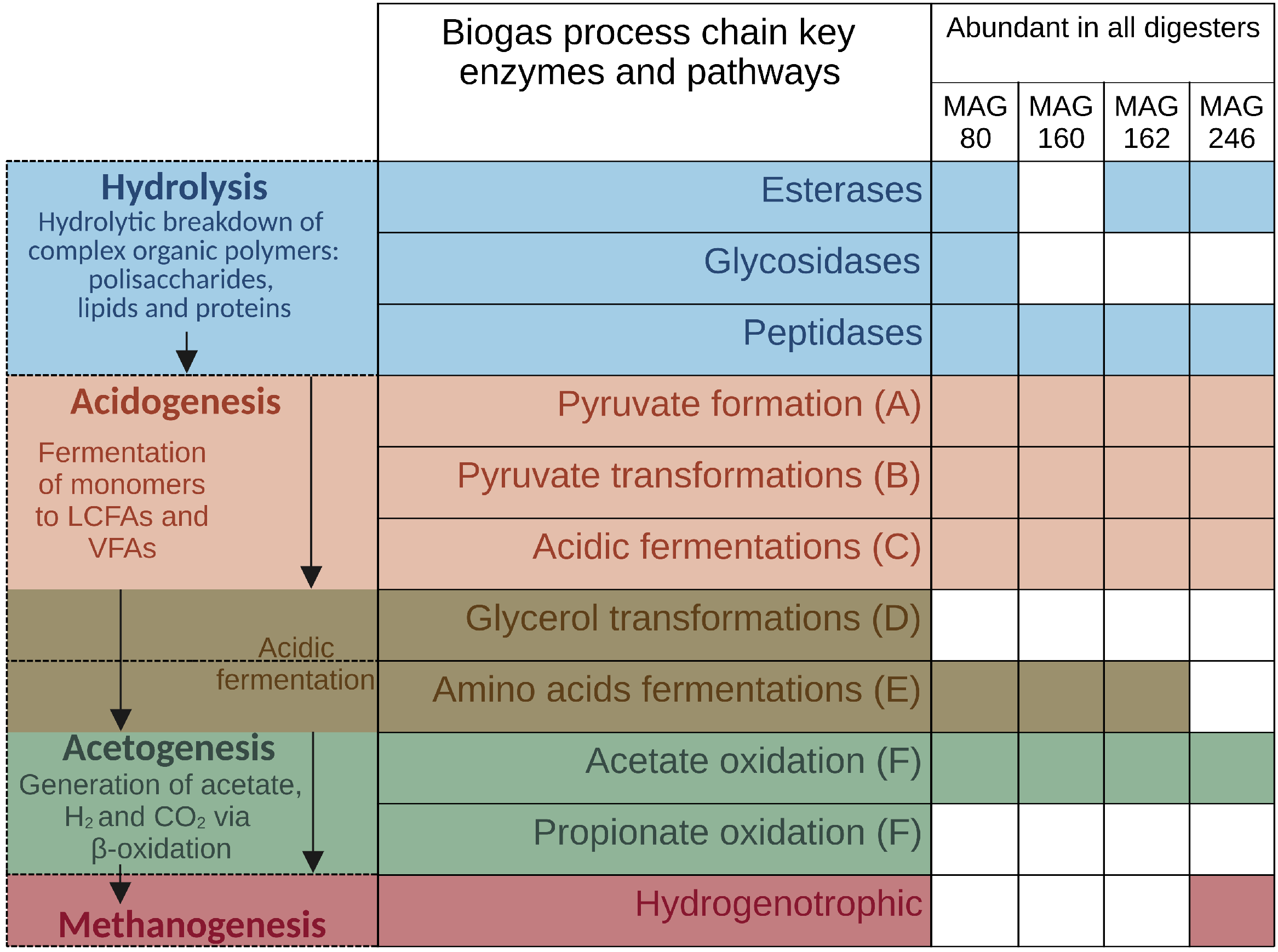
3.4. MAG-Centric Metagenome and Metaproteome Analyses Enabled Characterization of the Functional Potential, Expressed Functions and Role of Specific MAGs in the Biogas Process
3.4.1. Differentially Abundant High-Quality MAGs Showed Adaptations to the Different Process Conditions and Their Role in the Three Digesters Was Deduced Based on MAG-Centric Metaproteomics
MAGs Most Abundant and Active in Digester 1 (D1)
MAGs Most Abundant and Active in Digester 2 (D2)
MAGs Most Abundant and Active in Digester 3 (D3)
3.4.2. High-Quality MAGs with Similar Relative Abundances and Metabolic Activities in the Three Digesters Indicate Their Resilience and Importance for a Stable Biogas Process
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Theuerl, S.; Klang, J.; Prochnow, A. Process disturbances in agricultural biogas production—Causes, mechanisms and effects on the biogas microbiome: A review. Energies 2019, 12, 365. [Google Scholar] [CrossRef]
- De Vrieze, J.; Verstraete, W. Perspectives for microbial community composition in anaerobic digestion: From abundance and activity to connectivity. Environ. Microbiol. 2016, 18, 2797–2809. [Google Scholar] [CrossRef]
- Abendroth, C.; Hahnke, S.; Simeonov, C.; Klocke, M.; Casani-Miravalls, S.; Ramm, P.; Bürger, C.; Luschnig, O.; Porcar, M. Microbial communities involved in biogas production exhibit high resilience to heat shocks. Bioresour. Technol. 2018, 249, 1074–1079. [Google Scholar] [CrossRef]
- Hassa, J.; Maus, I.; Off, S.; Pühler, A.; Scherer, P.; Klocke, M.; Schlüter, A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl. Microbiol. Biotechnol. 2018, 102, 5045–5063. [Google Scholar] [CrossRef]
- Jünemann, S.; Kleinbölting, N.; Jaenicke, S.; Henke, C.; Hassa, J.; Nelkner, J.; Stolze, Y.; Albaum, S.P.; Schlüter, A.; Goesmann, A.; et al. Bioinformatics for NGS-based metagenomics and the application to biogas research. J. Biotechnol. 2017, 261, 10–23. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Kougias, P.G.; Luo, G.; Angelidaki, I. Metagenomic binning reveals the functional roles of core abundant microorganisms in twelve full-scale biogas plants. Water Res. 2018, 140, 123–134. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Rodriguez-R, L.M.; Kovalovszki, A.; Ziels, R.M.; Maus, I.; Zhu, X.; Kougias, P.G.; Basile, A.; Luo, G.; et al. New insights from the biogas microbiome by comprehensive genome-resolved metagenomics of nearly 1600 species originating from multiple anaerobic digesters. Biotechnol. Biofuels 2020, 13, 25. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Goesmann, A.; Jaenicke, S.; Jünemann, S.; Eikmeyer, F.; Szczepanowski, R.; Al-Soud, W.A.; Sørensen, S.; Pühler, A.; Schlüter, A. Profiling of the metabolically active community from a production-scale biogas plant by means of high-throughput metatranscriptome sequencing. J. Biotechnol. 2012, 158, 248–258. [Google Scholar] [CrossRef]
- Maus, I.; Koeck, D.E.; Cibis, K.G.; Hahnke, S.; Kim, Y.S.; Langer, T.; Kreubel, J.; Erhard, M.; Bremges, A.; Off, S.; et al. Unraveling the microbiome of a thermophilic biogas plant by metagenome and metatranscriptome analysis complemented by characterization of bacterial and archaeal isolates. Biotechnol. Biofuels 2016, 9, 171. [Google Scholar] [CrossRef]
- Maus, I.; Klocke, M.; Derenkó, J.; Stolze, Y.; Beckstette, M.; Jost, C.; Wibberg, D.; Blom, J.; Henke, C.; Willenbücher, K.; et al. Impact of process temperature and organic loading rate on cellulolytic/hydrolytic biofilm microbiomes during biomethanation of ryegrass silage revealed by genome-centered metagenomics and metatranscriptomics. Environ. Microbiome 2020, 15, 7. [Google Scholar] [CrossRef]
- Wirth, R.; Bagi, Z.; Shetty, P.; Szuhaj, M.; Cheung, T.T.S.; Kovács, K.L.; Maróti, G. Inter-kingdom interactions and stability of methanogens revealed by machine-learning guided multi-omics analysis of industrial-scale biogas plants. ISME J. 2023, 17, 1326–1339. [Google Scholar] [CrossRef]
- Hassa, J.; Wibberg, D.; Maus, I.; Pühler, A.; Schlüter, A. Genome analyses and genome-centered metatranscriptomics of Methanothermobacter wolfeii strain SIV6, isolated from a thermophilic production-scale biogas fermenter. Microorganisms 2019, 8, 13. [Google Scholar] [CrossRef]
- Maus, I.; Tubbesing, T.; Wibberg, D.; Heyer, R.; Hassa, J.; Tomazetto, G.; Huang, L.; Bunk, B.; Spröer, C.; Benndorf, D.; et al. The role of Petrimonas mucosa ING2-E5AT in mesophilic biogas reactor systems as deduced from multiomics analyses. Microorganisms 2020, 8, 2024. [Google Scholar] [CrossRef]
- Ortseifen, V.; Stolze, Y.; Maus, I.; Sczyrba, A.; Bremges, A.; Albaum, S.P.; Jaenicke, S.; Fracowiak, J.; Pühler, A.; Schlüter, A. An integrated metagenome and-proteome analysis of the microbial community residing in a biogas production plant. J. Biotechnol. 2016, 231, 268–279. [Google Scholar] [CrossRef]
- Buettner, C.; von Bergen, M.; Jehmlich, N.; Noll, M. Pseudomonas spp. are key players in agricultural biogas substrate degradation. Sci. Rep. 2019, 9, 12871. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Siewert, C.; Kohrs, F.; Greve, J.; Maus, I.; Klang, J.; Klocke, M.; Heiermann, M.; Hoffmann, M.; et al. Metaproteome analysis reveals that syntrophy, competition, and phage-host interaction shape microbial communities in biogas plants. Microbiome 2019, 7, 69. [Google Scholar] [CrossRef]
- Kohrs, F.; Heyer, R.; Magnussen, A.; Benndorf, D.; Muth, T.; Behne, A.; Rapp, E.; Kausmann, R.; Heiermann, M.; Klocke, M.; et al. Sample prefractionation with liquid isoelectric focusing enables in depth microbial metaproteome analysis of mesophilic and thermophilic biogas plants. Anaerobe 2014, 29, 59–67. [Google Scholar] [CrossRef]
- Heyer, R.; Kohrs, F.; Benndorf, D.; Rapp, E.; Kausmann, R.; Heiermann, M.; Klocke, M.; Reichl, U. Metaproteome analysis of the microbial communities in agricultural biogas plants. New Biotechnol. 2013, 30, 614–622. [Google Scholar] [CrossRef]
- Yue, Y.; Huang, H.; Qi, Z.; Dou, H.M.; Liu, X.Y.; Han, T.F.; Chen, Y.; Song, X.J.; Zhang, Y.H.; Tu, J. Evaluating metagenomics tools for genome binning with real metagenomic datasets and CAMI datasets. BMC Bioinform. 2020, 21, 334. [Google Scholar] [CrossRef]
- Yang, C.; Chowdhury, D.; Zhang, Z.; Cheung, W.K.; Lu, A.; Bian, Z.; Zhang, L. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Comput. Struct. Biotechnol. J. 2021, 19, 6301–6314. [Google Scholar] [CrossRef]
- Pereira, M.B.; Wallroth, M.; Jonsson, V.; Kristiansson, E. Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genom. 2018, 19, 274. [Google Scholar] [CrossRef]
- Hassa, J.; Klang, J.; Benndorf, D.; Pohl, M.; Hülsemann, B.; Mächtig, T.; Effenberger, M.; Pühler, A.; Schlüter, A.; Theuerl, S. Indicative Marker Microbiome Structures Deduced from the Taxonomic Inventory of 67 Full-Scale Anaerobic Digesters of 49 Agricultural Biogas Plants. Microorganisms 2021, 9, 1457. [Google Scholar] [CrossRef]
- Liebetrau, J. Collection of Measurement Methods for Biogas: Methods to Determine Parameters for Analysis Purposes and Parameters that Describe Processes in the Biogas Sector; DBFZ Deutsches Biomasseforschungszentrum Gemeinnützige GmbH: Leipzig, Germany, 2016. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 September 2023).
- Li, H. Seqtk: A Fast and Lightweight Tool for Processing FASTA or FASTQ Sequences. 2012. Available online: https://github.com/lh3/seqtk (accessed on 10 September 2023).
- Jaenicke, S.; Albaum, S.P.; Blumenkamp, P.; Linke, B.; Stoye, J.; Goesmann, A. Flexible metagenome analysis using the MGX framework. Microbiome 2018, 6, 1–9. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Manor, O.; Borenstein, E. MUSiCC: A marker genes based framework for metagenomic normalization and accurate profiling of gene abundances in the microbiome. Genome Biol. 2015, 16, 53. [Google Scholar] [CrossRef]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophor. Int. J. 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Technical Report; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Palù, M.; Basile, A.; Zampieri, G.; Treu, L.; Rossi, A.; Morlino, M.S.; Campanaro, S. KEMET–A python tool for KEGG Module evaluation and microbial genome annotation expansion. Comput. Struct. Biotechnol. J. 2022, 20, 1481–1486. [Google Scholar] [CrossRef]
- Sikora, A.; Detman, A.; Mielecki, D.; Chojnacka, A.; Błaszczyk, M. Searching for metabolic pathways of anaerobic digestion: A useful list of the key enzymes. In Anaerobic Digestion; IntechOpen Rijeka: Rijeka, Croatia, 2019; pp. 13–31. [Google Scholar]
- Calusinska, M.; Goux, X.; Fossépré, M.; Muller, E.E.; Wilmes, P.; Delfosse, P. A year of monitoring 20 mesophilic full-scale bioreactors reveals the existence of stable but different core microbiomes in bio-waste and wastewater anaerobic digestion systems. Biotechnol. Biofuels 2018, 11, 1–19. [Google Scholar] [CrossRef]
- Maus, I.; Kim, Y.S.; Wibberg, D.; Stolze, Y.; Off, S.; Antonczyk, S.; Pühler, A.; Scherer, P.; Schlüter, A. Biphasic study to characterize agricultural biogas plants by high-throughput 16S rRNA gene amplicon sequencing and microscopic analysis. J. Microbiol. Biotechnol. 2017, 27, 321–334. [Google Scholar] [CrossRef]
- Stolze, Y.; Bremges, A.; Rumming, M.; Henke, C.; Maus, I.; Pühler, A.; Sczyrba, A.; Schlüter, A. Identification and genome reconstruction of abundant distinct taxa in microbiomes from one thermophilic and three mesophilic production-scale biogas plants. Biotechnol. Biofuels 2016, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- De Vrieze, J.; Saunders, A.M.; He, Y.; Fang, J.; Nielsen, P.H.; Verstraete, W.; Boon, N. Ammonia and temperature determine potential clustering in the anaerobic digestion microbiome. Water Res. 2015, 75, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, C.; Al-Soud, W.A.; Larsson, M.; Alm, E.; Yekta, S.S.; Svensson, B.H.; Sørensen, S.J.; Karlsson, A. 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol. Ecol. 2013, 85, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, J.S.; de Jonge, N.; Macêdo, W.V.; Dalby, F.R.; Feilberg, A.; Nielsen, J.L. Characterisation of cellulose-degrading organisms in an anaerobic digester. Bioresour. Technol. 2022, 351, 126933. [Google Scholar]
- Ben Hania, W.; Godbane, R.; Postec, A.; Hamdi, M.; Ollivier, B.; Fardeau, M.L. Defluviitoga tunisiensis gen. nov., sp. nov., a thermophilic bacterium isolated from a mesothermic and anaerobic whey digester. Int. J. Syst. Evol. Microbiol. 2012, 62, 1377–1382. [Google Scholar] [CrossRef]
- Zhang, X.; Tu, B.; Dai, L.R.; Lawson, P.A.; Zheng, Z.Z.; Liu, L.Y.; Deng, Y.; Zhang, H.; Cheng, L. Petroclostridium xylanilyticum gen. nov., sp. nov., a xylan-degrading bacterium isolated from an oilfield, and reclassification of clostridial cluster III members into four novel genera in a new Hungateiclostridiaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 3197–3211. [Google Scholar] [CrossRef]
- Wasserfallen, A.; Nölling, J.; Pfister, P.; Reeve, J.; Conway de Macario, E. Phylogenetic analysis of 18 thermophilic Methanobacterium isolates supports the proposals to create a new genus, Methanothermobacter gen. nov., and to reclassify several isolates in three species, Methanothermobacter thermautotrophicus comb. nov., Methanothermobacter wolfeii comb. nov., and Methanothermobacter marburgensis sp. nov. Int. J. Syst. Evol. Microbiol. 2000, 50, 43–53. [Google Scholar]
- Tomazetto, G.; Hahnke, S.; Koeck, D.E.; Wibberg, D.; Maus, I.; Pühler, A.; Klocke, M.; Schlüter, A. Complete genome analysis of Clostridium bornimense strain M2/40T: A new acidogenic Clostridium species isolated from a mesophilic two-phase laboratory-scale biogas reactor. J. Biotechnol. 2016, 232, 38–49. [Google Scholar] [CrossRef]
- Kim, M.D.; Song, M.; Jo, M.; Shin, S.G.; Khim, J.H.; Hwang, S. Growth condition and bacterial community for maximum hydrolysis of suspended organic materials in anaerobic digestion of food waste-recycling wastewater. Appl. Microbiol. Biotechnol. 2010, 85, 1611–1618. [Google Scholar] [CrossRef]
- Ziganshin, A.M.; Liebetrau, J.; Pröter, J.; Kleinsteuber, S. Microbial community structure and dynamics during anaerobic digestion of various agricultural waste materials. Appl. Microbiol. Biotechnol. 2013, 97, 5161–5174. [Google Scholar] [CrossRef]
- Sun, L.; Liu, T.; Müller, B.; Schnürer, A. The microbial community structure in industrial biogas plants influences the degradation rate of straw and cellulose in batch tests. Biotechnol. Biofuels 2016, 9, 128. [Google Scholar] [CrossRef]
- Gabris, C.; Bengelsdorf, F.R.; Dürre, P. Analysis of the key enzymes of butyric and acetic acid fermentation in biogas reactors. Microb. Biotechnol. 2015, 8, 865–873. [Google Scholar] [CrossRef]
- Zverlov, V.V.; Hiegl, W.; Köck, D.E.; Kellermann, J.; Köllmeier, T.; Schwarz, W.H. Hydrolytic bacteria in mesophilic and thermophilic degradation of plant biomass. Eng. Life Sci. 2010, 10, 528–536. [Google Scholar] [CrossRef]
- Soutschek, E.; Winter, J.; Schindler, F.; Kandler, O. Acetomicrobium flavidum, gen. nov., sp. nov., a thermophilic, anaerobic bacterium from sewage sludge, forming acetate, CO2 and H2 from glucose. Syst. Appl. Microbiol. 1984, 5, 377–390. [Google Scholar] [CrossRef]
- Schlüter, A.; Bekel, T.; Diaz, N.N.; Dondrup, M.; Eichenlaub, R.; Gartemann, K.H.; Krahn, I.; Krause, L.; Krömeke, H.; Kruse, O.; et al. The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J. Biotechnol. 2008, 136, 77–90. [Google Scholar] [CrossRef]
- Sanjuán, R. Collective infectious units in viruses. Trends Microbiol. 2017, 25, 402–412. [Google Scholar] [CrossRef]
- Shan, J.; Korbsrisate, S.; Withatanung, P.; Adler, N.L.; Clokie, M.R.; Galyov, E.E. Temperature dependent bacteriophages of a tropical bacterial pathogen. Front. Microbiol. 2014, 5, 599. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, H.; Morita, M. Phage DNA packaging. Genes Cells 1997, 2, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Feiss, M.; Rao, V.B. The bacteriophage DNA packaging machine. Adv. Exp. Med. Biol. 2012, 726, 489–509. [Google Scholar] [PubMed]
- Clark, A.J. Progress toward a metabolic interpretation of genetic recombination of Escherichia coli and bacteriophage lambda. Genetics 1974, 78, 259. [Google Scholar] [CrossRef] [PubMed]
- Ashley, N.; Hurst, T.J. Acid and alkaline phosphatase activity in anaerobic digested sludge: A biochemical predictor of digester failure. Water Res. 1981, 15, 633–638. [Google Scholar] [CrossRef]
- Zhenglan, W.; Yue, G.; Lanying, L. A study of phosphatase activity in anaerobic sludge digestion. Water Res. 1990, 24, 917–920. [Google Scholar] [CrossRef]
- National Research Council. Nutrient Requirements of Dairy Cattle, 7th revised ed.; The National Academies Press: Washington, DC, USA, 2001. [Google Scholar]
- Javed, A.; Ahmad, A.; Tahir, A.; Shabbir, U.; Nouman, M.; Hameed, A. Potato Peel Waste—Its Nutraceutical, Industrial and Biotechnological Applacations. Aims Agric. Food 2019, 4, 807–823. [Google Scholar] [CrossRef]
- Bertoldo, C.; Antranikian, G. Starch-hydrolyzing enzymes from thermophilic archaea and bacteria. Curr. Opin. Chem. Biol. 2002, 6, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Achinas, S.; Zhao, J.; Geurkink, B.; Krooneman, J.; Euverink, G.J.W. Co-digestion of cow and sheep manure: Performance evaluation and relative microbial activity. Renew. Energy 2020, 153, 553–563. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, J.; Achinas, S.; Zhang, Z.; Krooneman, J.; Euverink, G.J.W. The biomethanation of cow manure in a continuous anaerobic digester can be boosted via a bioaugmentation culture containing Bathyarchaeota. Sci. Total Environ. 2020, 745, 141042. [Google Scholar] [CrossRef]
- Bah, H.; Zhang, W.; Wu, S.; Qi, D.; Kizito, S.; Dong, R. Evaluation of batch anaerobic co-digestion of palm pressed fiber and cattle manure under mesophilic conditions. Waste Manag. 2014, 34, 1984–1991. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, F.; Yu, J.; Cai, Y.; Luo, X.; Cui, Z.; Hu, Y.; Wang, X. Co-digestion of oat straw and cow manure during anaerobic digestion: Stimulative and inhibitory effects on fermentation. Bioresour. Technol. 2018, 269, 143–152. [Google Scholar] [CrossRef]
- Cestonaro, T.; de Mendonça Costa, M.S.S.; de Mendonça Costa, L.A.; Rozatti, M.A.T.; Pereira, D.C.; Lorin, H.E.F.; Carneiro, L.J. The anaerobic co-digestion of sheep bedding and ≥50% cattle manure increases biogas production and improves biofertilizer quality. Waste Manag. 2015, 46, 612–618. [Google Scholar] [CrossRef]
- Liao, W.; Wen, Z.; Hurley, S.; Liu, Y.; Liu, C.; Chen, S. Effects of hemicellulose and lignin on enzymatic hydrolysis of cellulose from dairy manure. Appl. Biochem. Biotechnol. 2005, 124, 1017–1030. [Google Scholar] [CrossRef]
- Khan, M.U.; Ahring, B.K. Improving the biogas yield of manure: Effect of pretreatment on anaerobic digestion of the recalcitrant fraction of manure. Bioresour. Technol. 2021, 321, 124427. [Google Scholar]
- Liu, M.; Bayjanov, J.R.; Renckens, B.; Nauta, A.; Siezen, R.J. The proteolytic system of lactic acid bacteria revisited: A genomic comparison. BMC Genom. 2010, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rohrwild, M.; Coux, O.; Huang, H.; Moerschell, R.P.; Yoo, S.J.; Seol, J.H.; Chung, C.H.; Goldberg, A.L. HslV-HslU: A novel ATP-dependent protease complex in Escherichia coli related to the eukaryotic proteasome. Proc. Natl. Acad. Sci. USA 1996, 93, 5808–5813. [Google Scholar] [CrossRef]
- Seol, J.H.; Yoo, S.J.; Shin, D.H.; Shim, Y.K.; Kang, M.S.; Goldberg, A.L.; Chung, C.H. The heat-shock protein HslVU from Escherichia coli is a protein-activated ATPase as well as an ATP-dependent proteinase. Eur. J. Biochem. 1997, 247, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Adghim, M.; Abdallah, M.; Saad, S.; Shanableh, A.; Sartaj, M. Assessment of the biochemical methane potential of mono-and co-digested dairy farm wastes. Waste Manag. Res. 2020, 38, 88–99. [Google Scholar] [CrossRef]
- Kelley, J.J.; Dekker, E.E. Identity of Escherichia coli D-1-amino-2-propanol: NAD+ oxidoreductase with E. coli glycerol dehydrogenase but not with Neisseria gonorrhoeae 1, 2-propanediol: NAD+ oxidoreductase. J. Bacteriol. 1985, 162, 170–175. [Google Scholar] [CrossRef]
- Subedi, K.P.; Kim, I.; Kim, J.; Min, B.; Park, C. Role of GldA in dihydroxyacetone and methylglyoxal metabolism of Escherichia coli K12. FEMS Microbiol. Lett. 2008, 279, 180–187. [Google Scholar] [CrossRef]
- Wang, Y.; Tao, F.; Xu, P. Glycerol dehydrogenase plays a dual role in glycerol metabolism and 2, 3-butanediol formation in Klebsiella pneumoniae. J. Biol. Chem. 2014, 289, 6080–6090. [Google Scholar] [CrossRef]
- Galushko, A.S.; Schink, B. Oxidation of acetate through reactions of the citric acid cycle by Geobacter sulfurreducens in pure culture and in syntrophic coculture. Arch. Microbiol. 2000, 174, 314–321. [Google Scholar] [CrossRef]
- Bertsch, J.; Siemund, A.L.; Kremp, F.; Müller, V. A novel route for ethanol oxidation in the acetogenic bacterium Acetobacterium woodii: The acetaldehyde/ethanol dehydrogenase pathway. Environ. Microbiol. 2016, 18, 2913–2922. [Google Scholar] [CrossRef]
- Schmidt, A.; Müller, N.; Schink, B.; Schleheck, D. A proteomic view at the biochemistry of syntrophic butyrate oxidation in Syntrophomonas wolfei. PLoS ONE 2013, 8, e56905. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Worm, P.; Schink, B.; Stams, A.J.; Plugge, C.M. Syntrophic butyrate and propionate oxidation processes: From genomes to reaction mechanisms. Environ. Microbiol. Rep. 2010, 2, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Teater, C.; MacLellan, J.; Liu, Y.; Liao, W. Development of a new bioethanol feedstock–anaerobically digested fiber from confined dairy operations using different digestion configurations. Biomass Bioenergy 2011, 35, 1946–1953. [Google Scholar] [CrossRef]
- Lim, J.W.; Park, T.; Tong, Y.W.; Yu, Z. The microbiome driving anaerobic digestion and microbial analysis. In Advances in Bioenergy; Elsevier: Amsterdam, The Netherlands, 2020; Volume 5, pp. 1–61. [Google Scholar]
- Hanreich, A.; Schimpf, U.; Zakrzewski, M.; Schlüter, A.; Benndorf, D.; Heyer, R.; Rapp, E.; Pühler, A.; Reichl, U.; Klocke, M. Metagenome and metaproteome analyses of microbial communities in mesophilic biogas-producing anaerobic batch fermentations indicate concerted plant carbohydrate degradation. Syst. Appl. Microbiol. 2013, 36, 330–338. [Google Scholar] [CrossRef]
- Lü, F.; Bize, A.; Guillot, A.; Monnet, V.; Madigou, C.; Chapleur, O.; Mazéas, L.; He, P.; Bouchez, T. Metaproteomics of cellulose methanisation under thermophilic conditions reveals a surprisingly high proteolytic activity. ISME J. 2014, 8, 88–102. [Google Scholar] [CrossRef]
- Ma, S.; Jiang, F.; Huang, Y.; Zhang, Y.; Wang, S.; Fan, H.; Liu, B.; Li, Q.; Yin, L.; Wang, H.; et al. A microbial gene catalog of anaerobic digestion from full-scale biogas plants. GigaScience 2021, 10, giaa164. [Google Scholar] [CrossRef]
- Heyer, R.; Kohrs, F.; Reichl, U.; Benndorf, D. Metaproteomics of complex microbial communities in biogas plants. Microb. Biotechnol. 2015, 8, 749–763. [Google Scholar] [CrossRef]
- Jacq-Bailly, A.; Benvenuti, M.; Payne, N.; Kpebe, A.; Felbek, C.; Fourmond, V.; Léger, C.; Brugna, M.; Baffert, C. Electrochemical characterization of a complex FeFe hydrogenase, the electron-bifurcating Hnd from Desulfovibrio fructosovorans. Front. Chem. 2021, 8, 573305. [Google Scholar] [CrossRef]
- Kpebe, A.; Benvenuti, M.; Guendon, C.; Rebai, A.; Fernandez, V.; Le Laz, S.; Etienne, E.; Guigliarelli, B.; García-Molina, G.; de Lacey, A.L.; et al. A new mechanistic model for an O2-protected electron-bifurcating hydrogenase, Hnd from Desulfovibrio fructosovorans. Biochim. Biophys. Acta (BBA)-Bioenerg. 2018, 1859, 1302–1312. [Google Scholar] [CrossRef]
- De Luca, G.; de Philip, P.; Rousset, M.; Belaich, J.P.; Dermoun, Z. The NADP-Reducing Hydrogenase ofDesulfovibrio fructosovorans: Evidence for a Native Complex with Hydrogen-Dependent Methyl-Viologen-Reducing Activity. Biochem. Biophys. Res. Commun. 1998, 248, 591–596. [Google Scholar] [CrossRef]
- Malki, S.; Saimmaime, I.; De Luca, G.; Rousset, M.; Dermoun, Z.; Belaich, J.P. Characterization of an operon encoding an NADP-reducing hydrogenase in Desulfovibrio fructosovorans. J. Bacteriol. 1995, 177, 2628–2636. [Google Scholar] [CrossRef] [PubMed]
- Constant, P.; Hallenbeck, P.C. Hydrogenase. In Biohydrogen; Elsevier: Amsterdam, The Netherlands, 2019; p. 69. [Google Scholar]
- Soboh, B.; Linder, D.; Hedderich, R. A multisubunit membrane-bound [NiFe] hydrogenase and an NADH-dependent Fe-only hydrogenase in the fermenting bacterium Thermoanaerobacter tengcongensis. Microbiology 2004, 150, 2451–2463. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Zeng, Y.; Wang, H.Z.; Chen, Y.T.; Tang, Y.Q. Proteiniphilum and Methanothrix harundinacea became dominant acetate utilizers in a methanogenic reactor operated under strong ammonia stress. Front. Microbiol. 2022, 13, 1098814. [Google Scholar] [CrossRef] [PubMed]
- Mei, R.; Nobu, M.K.; Narihiro, T.; Liu, W.T. Metagenomic and metatranscriptomic analyses revealed uncultured bacteroidales populations as the dominant proteolytic amino acid degraders in anaerobic digesters. Front. Microbiol. 2020, 11, 593006. [Google Scholar] [CrossRef]
- Koeck, D.E.; Mechelke, M.; Zverlov, V.V.; Liebl, W.; Schwarz, W.H. Herbivorax saccincola gen. nov., sp. nov., a cellulolytic, anaerobic, thermophilic bacterium isolated via in sacco enrichments from a lab-scale biogas reactor. Int. J. Syst. Evol. Microbiol. 2016, 66, 4458–4463. [Google Scholar] [CrossRef]
- Patel, G.; Khan, A.; Agnew, B.; Colvin, J. Isolation and characterization of an anaerobic, cellulolytic microorganism, Acetivibrio cellulolyticus gen. nov., sp. nov. Int. J. Syst. Evol. Microbiol. 1980, 30, 179–185. [Google Scholar] [CrossRef]
- Kakuk, B.; Wirth, R.; Maróti, G.; Szuhaj, M.; Rakhely, G.; Laczi, K.; Kovács, K.L.; Bagi, Z. Early response of methanogenic archaea to H2 as evaluated by metagenomics and metatranscriptomics. Microb. Cell Factories 2021, 20, 127. [Google Scholar] [CrossRef]
- Perman, E.; Schnürer, A.; Björn, A.; Moestedt, J. Serial anaerobic digestion improves protein degradation and biogas production from mixed food waste. Biomass Bioenergy 2022, 161, 106478. [Google Scholar] [CrossRef]
- Yokoyama, H.; Wagner, I.D.; Wiegel, J. Caldicoprobacter oshimai gen. nov., sp. nov., an anaerobic, xylanolytic, extremely thermophilic bacterium isolated from sheep faeces, and proposal of Caldicoprobacteraceae fam. nov. Int. J. Syst. Evol. Microbiol. 2010, 60, 67–71. [Google Scholar] [CrossRef]
- Zhang, J.; Loh, K.C.; Lee, J.; Wang, C.H.; Dai, Y.; Wah Tong, Y. Three-stage anaerobic co-digestion of food waste and horse manure. Sci. Rep. 2017, 7, 1269. [Google Scholar] [CrossRef]
- Cohn, M.T.; Ingmer, H.; Mulholland, F.; Jørgensen, K.; Wells, J.M.; Brøndsted, L. Contribution of conserved ATP-dependent proteases of Campylobacter jejuni to stress tolerance and virulence. Appl. Environ. Microbiol. 2007, 73, 7803–7813. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Qian, Y.; Zhou, Z.; Ertmer, S.; Vivas, E.I.; Lan, F.; Hamilton, J.J.; Rey, F.E.; Anantharaman, K.; Venturelli, O.S. Polysaccharide utilization loci in Bacteroides determine population fitness and community-level interactions. Cell Host Microbe 2022, 30, 200–215. [Google Scholar] [CrossRef]
- Grondin, J.M.; Tamura, K.; Déjean, G.; Abbott, D.W.; Brumer, H. Polysaccharide utilization loci: Fueling microbial communities. J. Bacteriol. 2017, 199, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Hahnke, S.; Maus, I.; Wibberg, D.; Tomazetto, G.; Pühler, A.; Klocke, M.; Schlüter, A. Complete genome sequence of the novel Porphyromonadaceae bacterium strain ING2-E5B isolated from a mesophilic lab-scale biogas reactor. J. Biotechnol. 2015, 193, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Campanaro, S.; Treu, L.; Kougias, P.G.; De Francisci, D.; Valle, G.; Angelidaki, I. Metagenomic analysis and functional characterization of the biogas microbiome using high throughput shotgun sequencing and a novel binning strategy. Biotechnol. Biofuels 2016, 9, 26. [Google Scholar] [CrossRef]
- Schnürer, A. Biogas production: Microbiology and technology. Adv. Biochem. Eng. Biotechnol. 2016, 156, 195–234. [Google Scholar]
- Zhu, N.; Yang, J.; Ji, L.; Liu, J.; Yang, Y.; Yuan, H. Metagenomic and metaproteomic analyses of a corn stover-adapted microbial consortium EMSD5 reveal its taxonomic and enzymatic basis for degrading lignocellulose. Biotechnol. Biofuels 2016, 9, 2343. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Yang, Y.-H. Microbial production of volatile fatty acids: Current status and future perspectives. Rev. Environ. Sci. Bio/Technol. 2017, 16, 327–345. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Coorevits, A.; Dinsdale, A.E.; Halket, G.; Lebbe, L.; De Vos, P.; Van Landschoot, A.; Logan, N.A. Taxonomic revision of the genus Geobacillus: Emendation of Geobacillus, G. stearothermophilus, G. jurassicus, G. toebii, G. thermodenitrificans and G. thermoglucosidans (nom. corrig., formerly ‘thermoglucosidasius’); transfer of Bacillus thermantarcticus to the genus as G. thermantarcticus comb. nov.; proposal of Caldibacillus debilis gen. nov., comb. nov.; transfer of G. tepidamans to Anoxybacillus as A. tepidamans comb. nov.; and proposal of Anoxybacillus caldiproteolyticus sp. nov. Int. J. Syst. Evol. Microbiol. 2012, 62, 1470–1485. [Google Scholar]
- Kotrba, P.; Inui, M.; Yukawa, H. Bacterial phosphotransferase system (PTS) in carbohydrate uptake and control of carbon metabolism. J. Biosci. Bioeng. 2001, 92, 502–517. [Google Scholar] [CrossRef]
- Schöcke, L.; Schink, B. Energetics and biochemistry of fermentative benzoate degradation by Syntrophus gentianae. Arch. Microbiol. 1999, 171, 331–337. [Google Scholar] [CrossRef][Green Version]
- Nakhate, S.; Gupta, R.; Poddar, B.; Singh, A.; Tikariha, H.; Pandit, P.; Khardenavis, A.; Purohit, H. Influence of lignin level of raw material on anaerobic digestion process in reorganization and performance of microbial community. Int. J. Environ. Sci. Technol. 2021, 19, 1819–1836. [Google Scholar] [CrossRef]
- Kumari, S.; Das, S. Bacterial enzymatic degradation of recalcitrant organic pollutants: Catabolic pathways and genetic regulations. Environ. Sci. Pollut. Res. 2023, 30, 79676–79705. [Google Scholar] [CrossRef]
- Chen, H.; Yang, T.; Shen, Z.; Yang, E.; Liu, K.; Wang, H.; Chen, J.; Sanjaya, E.H.; Wu, S. Can digestate recirculation promote biohythane production from two-stage co-digestion of rice straw and pig manure? J. Environ. Manag. 2022, 319, 115655. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, D.; Woese, C.R.; Bryant, M.P. Assignment of Fatty Acid-ß-Oxidizing Syntrophic Bacteria to Syntrophomonadaceae fam. nov. on the Basis of 16S rRNA Sequence Analyses. Int. J. Syst. Bacteriol. 1993, 43, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Hanreich, A.; Heyer, R.; Benndorf, D.; Rapp, E.; Pioch, M.; Reichl, U.; Klocke, M. Metaproteome analysis to determine the metabolically active part of a thermophilic microbial community producing biogas from agricultural biomass. Can. J. Microbiol. 2012, 58, 917–922. [Google Scholar] [CrossRef]
- Gaspari, M.; Treu, L.; Zhu, X.; Palù, M.; Angelidaki, I.; Campanaro, S.; Kougias, P.G. Microbial dynamics in biogas digesters treating lipid-rich substrates via genome-centric metagenomics. Sci. Total Environ. 2021, 778, 146296. [Google Scholar] [CrossRef] [PubMed]
- Tsapekos, P.; Treu, L.; Campanaro, S.; Centurion, V.B.; Zhu, X.; Peprah, M.; Zhang, Z.; Kougias, P.G.; Angelidaki, I. Pilot-scale biomethanation in a trickle bed reactor: Process performance and microbiome functional reconstruction. Energy Convers. Manag. 2021, 244, 114491. [Google Scholar] [CrossRef]
- Ebrahimian, F.; De Bernardini, N.; Tsapekos, P.; Treu, L.; Zhu, X.; Campanaro, S.; Karimi, K.; Angelidaki, I. Effect of pressure on biomethanation process and spatial stratification of microbial communities in trickle bed reactors under decreasing gas retention time. Bioresour. Technol. 2022, 361, 127701. [Google Scholar] [CrossRef]
- De Bernardini, N.; Basile, A.; Zampieri, G.; Kovalovszki, A.; De Diego Diaz, B.; Offer, E.; Wongfaed, N.; Angelidaki, I.; Kougias, P.G.; Campanaro, S.; et al. Integrating metagenomic binning with flux balance analysis to unravel syntrophies in anaerobic CO2 methanation. Microbiome 2022, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Meckenstock, R.U.; Boll, M.; Mouttaki, H.; Koelschbach, J.S.; Cunha Tarouco, P.; Weyrauch, P.; Dong, X.; Himmelberg, A.M. Anaerobic degradation of benzene and polycyclic aromatic hydrocarbons. Microb. Physiol. 2016, 26, 92–118. [Google Scholar] [CrossRef] [PubMed]
- Sim, Y.B.; Yang, J.; Joo, H.H.; Jung, J.H.; Kim, D.H.; Kim, S.H. Effect of bioaugmentation using Clostridium butyricum on the start-up and the performance of continuous biohydrogen production. Bioresour. Technol. 2022, 366, 128181. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biogas Digester | Maize Silage [%] | Grass Silage [%] | Cereals [%] | Solid Manure (Sheep, Cattle) [%] | Chicken Manure [%] | Potato Peels [%] | OLR [kgVS md] | HRT [d] | Temperature [°C] |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 34.00 | 27.85 | 6.99 | 25.38 | 5.8 | - | 4.53 | 71 | 44.5 |
| 2 | 36.20 | 29.45 | 7.39 | 21.96 | 5.0 | - | 4.32 | 75 | 50.0 |
| 3 | - | 20.55 | - | 59.32 | - | 20.13 | 0.41 | 475 | 56.3 |
| Contigs, Low Quality and HQ MAGs | Digester 1 | Digester 2 | Digester 3 | |
|---|---|---|---|---|
| Metagenome- based relative abundance | Unbinned contigs and low quality MAGs | 42.02% | 37.02% | 37.81% |
| HQ MAGs < 0.5% relative abundance | 11.77% | 6.48% | 5.85% | |
| HQ MAGs > 0.5% relative abundance | 46.21% | 56.50% | 56.34% | |
| Fraction of assigned metaproteins | Unbinned contigs and low quality MAGs | 35.03% | 36.06% | 44.19% |
| HQ MAGs < 0.5% relative abundance | 6.92% | 3.98% | 3.69% | |
| HQ MAGs > 0.5% relative abundance | 58.05% | 59.96% | 52.12% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassa, J.; Tubbesing, T.J.; Maus, I.; Heyer, R.; Benndorf, D.; Effenberger, M.; Henke, C.; Osterholz, B.; Beckstette, M.; Pühler, A.; et al. Uncovering Microbiome Adaptations in a Full-Scale Biogas Plant: Insights from MAG-Centric Metagenomics and Metaproteomics. Microorganisms 2023, 11, 2412. https://doi.org/10.3390/microorganisms11102412
Hassa J, Tubbesing TJ, Maus I, Heyer R, Benndorf D, Effenberger M, Henke C, Osterholz B, Beckstette M, Pühler A, et al. Uncovering Microbiome Adaptations in a Full-Scale Biogas Plant: Insights from MAG-Centric Metagenomics and Metaproteomics. Microorganisms. 2023; 11(10):2412. https://doi.org/10.3390/microorganisms11102412
Chicago/Turabian StyleHassa, Julia, Tom Jonas Tubbesing, Irena Maus, Robert Heyer, Dirk Benndorf, Mathias Effenberger, Christian Henke, Benedikt Osterholz, Michael Beckstette, Alfred Pühler, and et al. 2023. "Uncovering Microbiome Adaptations in a Full-Scale Biogas Plant: Insights from MAG-Centric Metagenomics and Metaproteomics" Microorganisms 11, no. 10: 2412. https://doi.org/10.3390/microorganisms11102412
APA StyleHassa, J., Tubbesing, T. J., Maus, I., Heyer, R., Benndorf, D., Effenberger, M., Henke, C., Osterholz, B., Beckstette, M., Pühler, A., Sczyrba, A., & Schlüter, A. (2023). Uncovering Microbiome Adaptations in a Full-Scale Biogas Plant: Insights from MAG-Centric Metagenomics and Metaproteomics. Microorganisms, 11(10), 2412. https://doi.org/10.3390/microorganisms11102412