



Application of Cloning-Free Genome Engineering to Escherichia coli

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Plasmids, Genes, and Cloning Procedures
2.3. PCR and Sanger Sequencing
2.4. Cloning-Free Genome Editing
2.5. Genomic DNA Extraction and MinION Nanopore Sequencing
2.6. Genome Assembly, Annotation, and Analyses
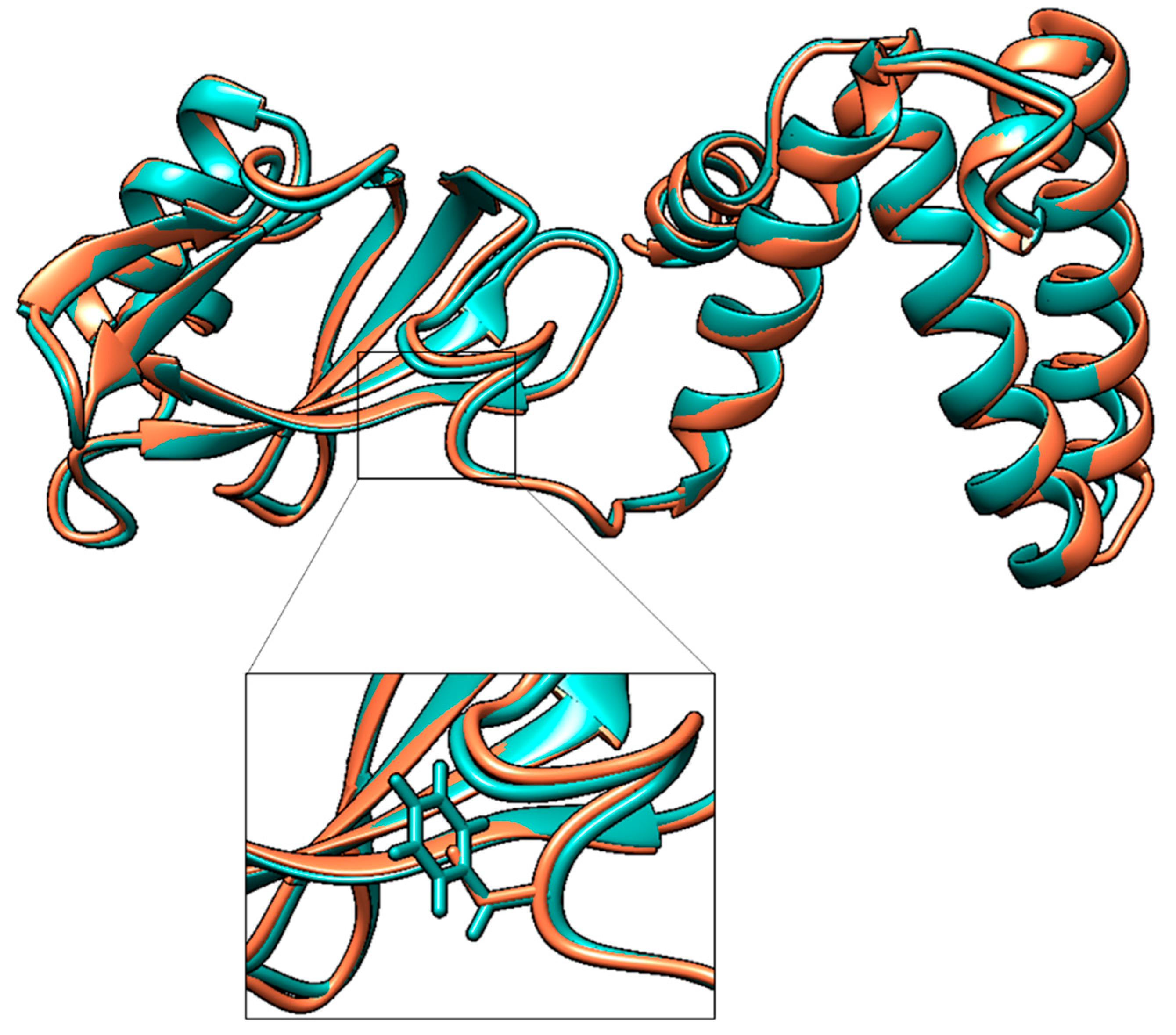
2.7. Prediction of Protein Three-Dimensional Structure
2.8. Isolation of HisI+ Revertants
- E. coli FB181 cells were grown overnight at 37 °C with shaking (150 rpm) in minimal medium Davis (MMD) [26] with glucose 1% and histidine 25 μg/mL.
- The optical density (O.D.600) of the culture was measured and the culture was diluted to O.D.600 0.1 in a final volume of 50 mL of MMD containing glucose 1% and histidine 25 μg/mL.
- The culture was then incubated at 37 °C with shaking (150 rpm). At the end of the log phase, cells were centrifuged, washed twice in saline solution (NaCl 0.9% w/v), and then spread on 100 mL MMD plates containing agar 1.6% and glucose 1% in the absence of histidine (three plates), or in the presence of histidine 0.3 μg/mL (three plates) or 1 μg/mL (three plates). An amount of 100 μL of 10−5 and 10−6 dilutions were plated on LB agar [25] to evaluate the cells’ vital titer.
- Vital titer plates were incubated at 37 °C overnight. Selective pressure plates were incubated at 37 °C for 15 days, and the appearance of HisI+ revertants was checked daily.
- HisI+ revertants were tested for their ability to grow in the absence or in low concentrations of histidine through streaking on MMD plates containing glucose 1% and histidine 0, 0.3, 1 μg/mL.
3. Results
3.1. CFGE of E. coli Wild-Type hisF Gene in E. coli FB182
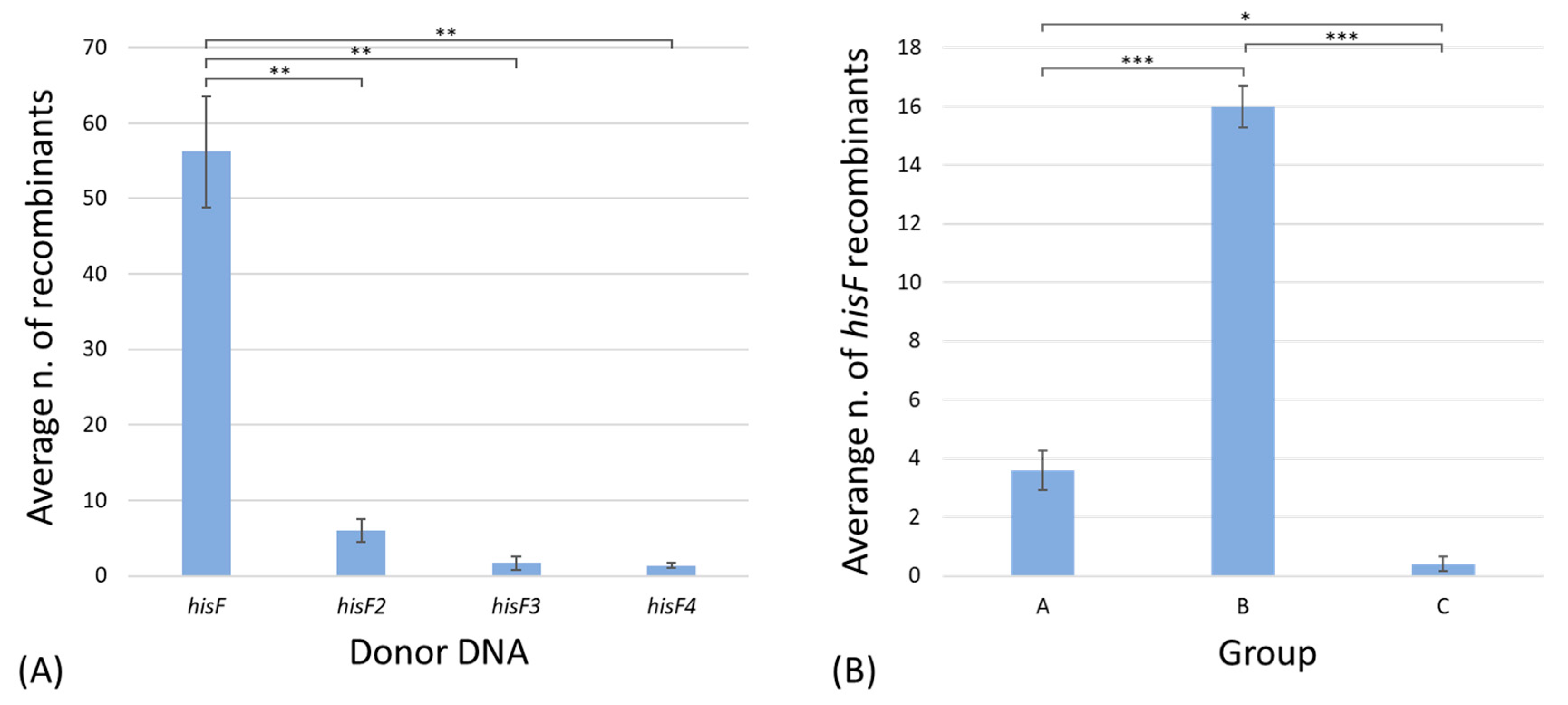
- Seven revertants owned a wild-type hisF gene replacing the E. coli FB182 mutated one (group A). Based on the previous assumption (i.e., the very low probability, less than 4%, of spontaneous restoring of the correct sequence) [21], they should be the result of a recombination event involving a single copy of the wild-type hisF gene;
- Thirty-two revertants possessed two or more in tandem hisF copies (group B) and are the result of recombinational events involving one, two, or more copies of the donor DNA;
- Just one transformant was a chromosomal revertant owning a hisF gene with a restored frame but a different sequence from the wild-type one (group C), according to Del Duca et al. [21].
- In E. coli FB182, only one copy of each his genes was found, located inside the compact E. coli his operon, and harboring, as expected, the single nucleotide deletion in position 719 of hisF.
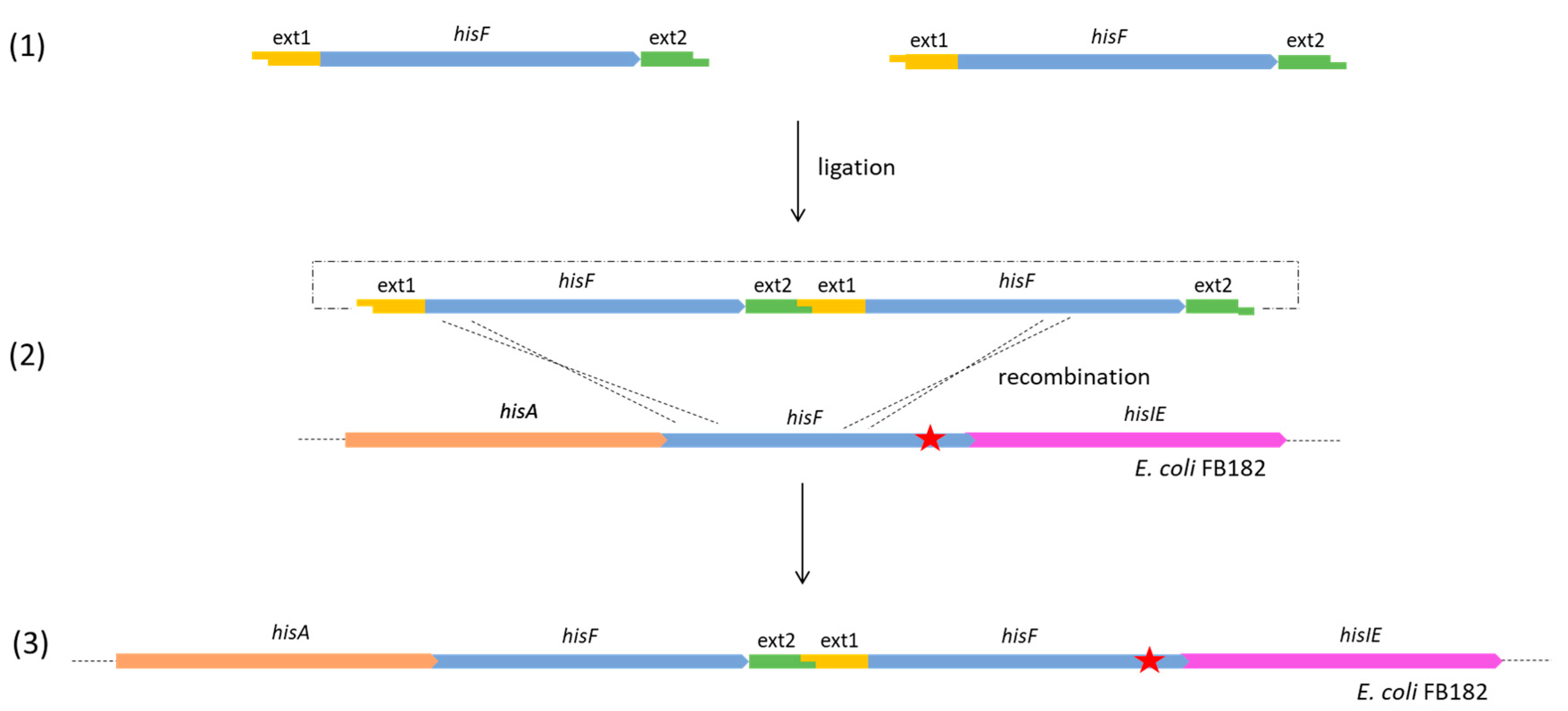
- For the clones belonging to group B, the complete genome analysis confirmed the presence of two in tandem hisF copies for 40_E1 and 50_E2, and three in tandem copies for 55_E1 and 20_E2. The analysis of the sequences allowed us to verify that, in all four clones, the last hisF copy carries the E. coli FB182 single nucleotide deletion, while the other one/two copies correspond to wild-type hisF. Moreover, the region between the in tandem hisF copies consists of the pGEM-T Easy vector region comprised between the EcoRI restriction sites and the TA cloning insertion site. This finding can be explained as follows: during the ligation step, two or more hisF copies joined each other with their EcoRI overhanging ends and then recombined with the E. coli FB182 hisF gene, as shown in Figure 3.
- No additional hisF gene or part thereof was found outside the his genomic locus in any of the four HisF+ revertants analyzed.
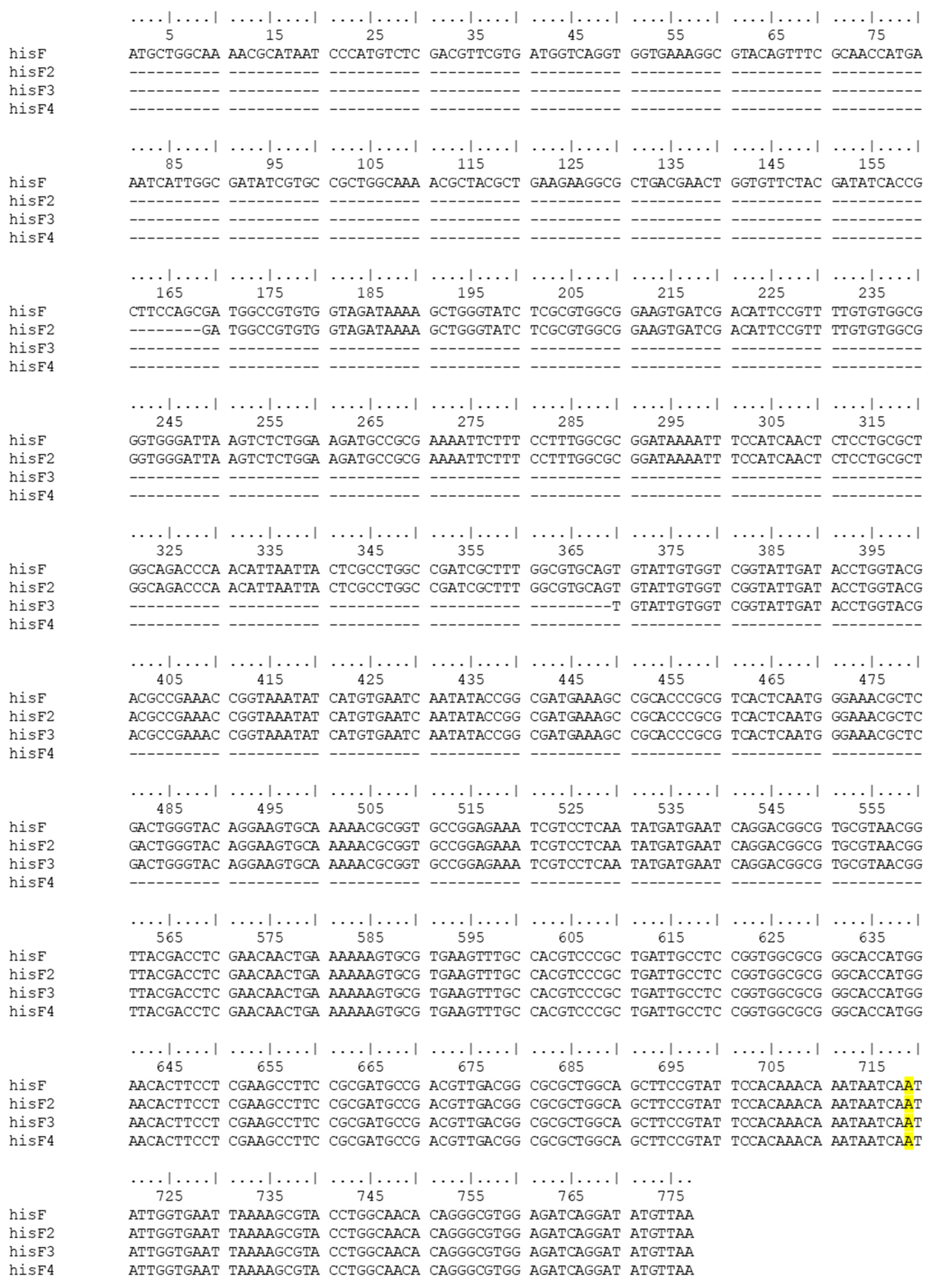
3.2. CFGE of E. coli Wild-Type hisF Gene Fragments in E. coli FB182
3.3. CFGE of E. coli Wild-Type hisIE Gene in E. coli FB181
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomas, C.M.; Nielsen, K.M. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 2005, 3, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Rivera, M.C.; Lake, J.A. Horizontal gene transfer among genomes: The complexity hypothesis. Proc. Natl. Acad. Sci. USA 1999, 96, 3801–3806. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M.; Van Der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz, F.; Davies, J. Horizontal gene transfer and the origin of species: Lessons from bacteria. Trends Microbiol. 2000, 8, 128–133. [Google Scholar] [CrossRef]
- Didelot, X.; Maiden, M.C.J. Impact of recombination on bacterial evolution. Trends Microbiol. 2010, 18, 315–322. [Google Scholar] [CrossRef]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef]
- Emamalipour, M.; Seidi, K.; Zununi Vahed, S.; Jahanban-Esfahlan, A.; Jaymand, M.; Majdi, H.; Amoozgar, Z.; Chitkushev, L.T.; Javaheri, T.; Jahanban-Esfahlan, R.; et al. Horizontal gene transfer: From evolutionary flexibility to disease progression. Front. Cell Dev. Biol. 2020, 8, 229. [Google Scholar] [CrossRef]
- Arnold, B.J.; Huang, I.T.; Hanage, W.P. Horizontal gene transfer and adaptive evolution in bacteria. Nat. Rev. Microbiol. 2021, 20, 206–218. [Google Scholar] [CrossRef]
- Faddetta, T.; Vassallo, A.; Del Duca, S.; Gallo, G.; Fani, R.; Puglia, A.M. Unravelling the DNA sequences carried by Streptomyces coelicolor membrane vesicles. Sci. Rep. 2022, 12, 1–8. [Google Scholar] [CrossRef]
- Johnston, C.; Martin, B.; Fichant, G.; Polard, P.; Claverys, J.P. Bacterial transformation: Distribution, shared mechanisms and divergent control. Nat. Rev. Microbiol. 2014, 12, 181–196. [Google Scholar] [CrossRef]
- Chan, W.T.; Verma, C.S.; Lane, D.P.; Gan, S.K.E. A comparison and optimization of methods and factors affecting the transformation of Escherichia coli. Biosci. Rep. 2013, 33, e00086. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.; Wood, E.A.; Romero, Z.J.; Cox, M.M. RecA-independent recombination: Dependence on the Escherichia coli RarA protein. Mol. Microbiol. 2021, 115, 1122–1137. [Google Scholar] [CrossRef]
- Kowalczykowski, S.C.; Dixon, D.A.; Eggleston, A.K.; Lauder, S.D.; Rehrauer, W.M. Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 1994, 58, 401–465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Buchholz, F.; Muyrers, J.P.P.; Francis Stewart, A. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 1998, 20, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef]
- Yu, B.J.; Kang, K.H.; Lee, J.H.; Sung, B.H.; Kim, M.S.; Kim, S.C. Rapid and efficient construction of markerless deletions in the Escherichia coli genome. Nucleic Acids Res. 2008, 36, e84. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Murphy, K.C.; Campellone, K.G.; Poteete, A.R. PCR-mediated gene replacement in Escherichia coli. Gene 2000, 246, 321–330. [Google Scholar] [CrossRef]
- Esvelt, K.M.; Wang, H.H. Genome-scale engineering for systems and synthetic biology. Mol. Syst. Biol. 2013, 9, 641. [Google Scholar] [CrossRef]
- Döhlemann, J.; Brennecke, M.; Becker, A. Cloning-free genome engineering in Sinorhizobium meliloti advances applications of Cre/loxP site-specific recombination. J. Biotechnol. 2016, 233, 160–170. [Google Scholar] [CrossRef]
- Del Duca, S.; Puglia, A.M.; Calderone, V.; Bazzicalupo, M.; Fani, R. Effect of Non-Lethal Selection on Spontaneous Revertants of Frameshift Mutations: The Escherichia coli hisF Case. Microorganisms 2022, 10, 692. [Google Scholar] [CrossRef] [PubMed]
- Kasai, T. Regulation of the expression of the histidine operon in Salmonella typhimurium. Nature 1974, 249, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, E.P.; Cater, M.S.; Matney, T.S.; Butler, M.A.; Greene, A. Genetic analysis of the histidine operon in Escherichia coli K12. Genetics 1970, 66, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Chioccioli, S.; Bogani, P.; Del Duca, S.; Castronovo, L.M.; Vassallo, A.; Puglia, A.M.; Fani, R. In vivo evaluation of the interaction between the Escherichia coli IGP synthase subunits using the Bacterial Two-Hybrid system. FEMS Microbiol. Lett. 2020, 367, 112. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989; ISBN 0879693096. [Google Scholar]
- Davis, B.D.; Mingioli, E.S. Mutants of Escherichia coli requiring methionine or vitamin B12. J. Bacteriol. 1950, 60, 17–28. [Google Scholar] [CrossRef]
- Hall, T.A. BIOEDIT: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Semenzato, G.; Alonso-Vásquez, T.; Del Duca, S.; Vassallo, A.; Riccardi, C.; Zaccaroni, M.; Mucci, N.; Padula, A.; Emiliani, G.; Piccionello, A.P.; et al. Genomic analysis of endophytic bacillus-related strains isolated from the medicinal plant Origanum vulgare L. revealed the presence of metabolic pathways involved in the biosynthesis of bioactive compounds. Microorganisms 2022, 10, 919. [Google Scholar] [CrossRef]
- Babraham Bioinformatics-FastQC A Quality Control tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 December 2022).
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Chilton, J.M.; Grüning, B.; Johnson, J.E.; Soranzo, N. NCBI BLAST+ integrated into Galaxy. Gigascience 2015, 4, s13742-015-0080-7. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Fani, R.; Liò, P.; Lazcano, A. Molecular evolution of the histidine biosynthetic pathway. J. Mol. Evol. 1995, 41, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, F.; Nie, Y.; Shang, G.; Zhang, H. Structural analysis of Shigella flexneri bi-functional enzyme HisIE in histidine biosynthesis. Biochem. Biophys. Res. Commun. 2019, 516, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Huang, H.V. Homologous recombination in Escherichia coli: Dependence on substrate length and homology. Genetics 1986, 112, 441–457. [Google Scholar] [CrossRef] [PubMed]
- King, S.R.; Richardson, J.P. Role of homology and pathway specificity for recombination between plasmids and bacteriophage lambda. Mol. Gen. Genet. 1986, 204, 141–147. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin/Heidelberg, Germany, 1970. [Google Scholar] [CrossRef]
- Fani, R.; Fondi, M. Origin and evolution of metabolic pathways. Phys. Life Rev. 2009, 6, 23–52. [Google Scholar] [CrossRef]
- Del Duca, S.; Chioccioli, S.; Vassallo, A.; Castronovo, L.M.; Fani, R. The role of gene elongation in the evolution of histidine biosynthetic genes. Microorganisms 2020, 8, 732. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′-3′) | Target Sequence | Amplicon |
|---|---|---|---|
| coli_hisF FW | ATGCTGGCAAAACGCATAA | E. coli hisF gene | hisF-777 bp hisF2-609 bp hisF3-408 bp hisF4-217 bp |
| coli_hisF_2 FW | GATGGCCGTGTGGTAGAT | E. coli hisF gene | |
| coli_hisF_3 FW | TGTATTGTGGTCGGTATTG | E. coli hisF gene | |
| coli_hisF_4 FW | TTACGACCTCGAACAACTG | E. coli hisF gene | |
| coli_hisF REV | TTAACATATCCTGATCTCCA | E. coli hisF gene | |
| coli_hisF_ext FW | GCGGCGTAATAGTTGGTCG | External to E. coli hisF gene | 959 bp |
| coli_hisF_ext REV | TCTAAGGCTTCCGGGTTCAT | External to E. coli hisF gene | |
| coli_hisIE_ext FW | GCACCATGGAACACTTCCTC | External to E. coli hisIE gene | 869 bp |
| coli_hisIE_ext REV | TACGCAATTACAACGCGAAG | External to E. coli hisIE gene | |
| M13 FW | GTAAAACGACGGCCAG | External to pGEM-T Easy MCS | variable |
| M13 REV | CAGGAAACAGCTATGAC | External to pGEM-T Easy MCS |
| Experiment | Donor DNA | |||
|---|---|---|---|---|
| hisF | hisF2 | hisF3 | hisF4 | |
| 777 bp | 609 bp | 408 bp | 217 bp | |
| 1 | 56 | - | - | - |
| 2 | 65 | - | - | - |
| 3 | 29 | 8 | 0 | 2 |
| 4 | 58 | 3 | 2 | 1 |
| 5 | 73 | 7 | 3 | 1 |
| Total amount of His+ colonies | 281 | 18 | 5 | 4 |
| Mean | 56.2 | 6 | 1.7 | 1.3 |
| Colony ID | Experiment | Sample Group | N. Contigs | Largest Contig (bp) | Total Length (bp) | N50 |
|---|---|---|---|---|---|---|
| FB182 | - | - | 1 | 4,637,980 | 4,637,980 | 4,637,980 |
| 40_E1 | 1 | B | 2 | 4,693,591 | 4,747,989 | 4,693,591 |
| 50_E2 | 2 | B | 1 | 4,666,291 | 4,666,291 | 4,666,291 |
| 55_E1 | 1 | B | 2 | 4,650,149 | 4,703,046 | 4,650,149 |
| 20_E2 | 2 | B | 2 | 4,645,202 | 4,745,589 | 4,645,202 |
| Experiment | Fragment | N. of His+ Colonies | N. of Characterized Colonies | Group A (1 hisF Copy) | Group B (2 or More hisF Copies) | Group C (Chromosomal Revertants) | |
|---|---|---|---|---|---|---|---|
| Name | Size (bp) | ||||||
| 1 | hisF | 777 | 56 | 20 | 3 | 17 | 0 |
| 2 | hisF | 777 | 65 | 20 | 4 | 15 | 1 |
| 3 | hisF | 777 | 29 | 20 | 3 | 16 | 1 |
| hisF2 | 609 | 8 | 8 | 5 | 3 | 0 | |
| hisF3 | 408 | 0 | 0 | 0 | 0 | 0 | |
| hisF4 | 217 | 2 | 2 | 1 | 0 | 1 | |
| 4 | hisF | 777 | 58 | 20 | 6 | 14 | 0 |
| hisF2 | 609 | 3 | 3 | 0 | 2 | 1 | |
| hisF3 | 408 | 2 | 2 | 0 | 1 | 1 | |
| hisF4 | 217 | 1 | 1 | 0 | 0 | 1 | |
| 5 | hisF | 777 | 73 | 20 | 2 | 18 | 0 |
| hisF2 | 609 | 7 | 7 | 1 | 6 | 0 | |
| hisF3 | 408 | 3 | 3 | 0 | 0 | 3 | |
| hisF4 | 217 | 1 | 1 | 0 | 0 | 1 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romeo, L.; Esposito, A.; Bernacchi, A.; Colazzo, D.; Vassallo, A.; Zaccaroni, M.; Fani, R.; Del Duca, S. Application of Cloning-Free Genome Engineering to Escherichia coli. Microorganisms 2023, 11, 215. https://doi.org/10.3390/microorganisms11010215
Romeo L, Esposito A, Bernacchi A, Colazzo D, Vassallo A, Zaccaroni M, Fani R, Del Duca S. Application of Cloning-Free Genome Engineering to Escherichia coli. Microorganisms. 2023; 11(1):215. https://doi.org/10.3390/microorganisms11010215
Chicago/Turabian StyleRomeo, Lucia, Antonia Esposito, Alberto Bernacchi, Daniele Colazzo, Alberto Vassallo, Marco Zaccaroni, Renato Fani, and Sara Del Duca. 2023. "Application of Cloning-Free Genome Engineering to Escherichia coli" Microorganisms 11, no. 1: 215. https://doi.org/10.3390/microorganisms11010215
APA StyleRomeo, L., Esposito, A., Bernacchi, A., Colazzo, D., Vassallo, A., Zaccaroni, M., Fani, R., & Del Duca, S. (2023). Application of Cloning-Free Genome Engineering to Escherichia coli. Microorganisms, 11(1), 215. https://doi.org/10.3390/microorganisms11010215