
Biogeographic Pattern and Network of Rhizosphere Fungal and Bacterial Communities in Panicum miliaceum Fields: Roles of Abundant and Rare Taxa


Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Site and Data Collection
2.2. DNA Extraction and Sequence Analysis
2.3. Statistical Analysis
3. Results
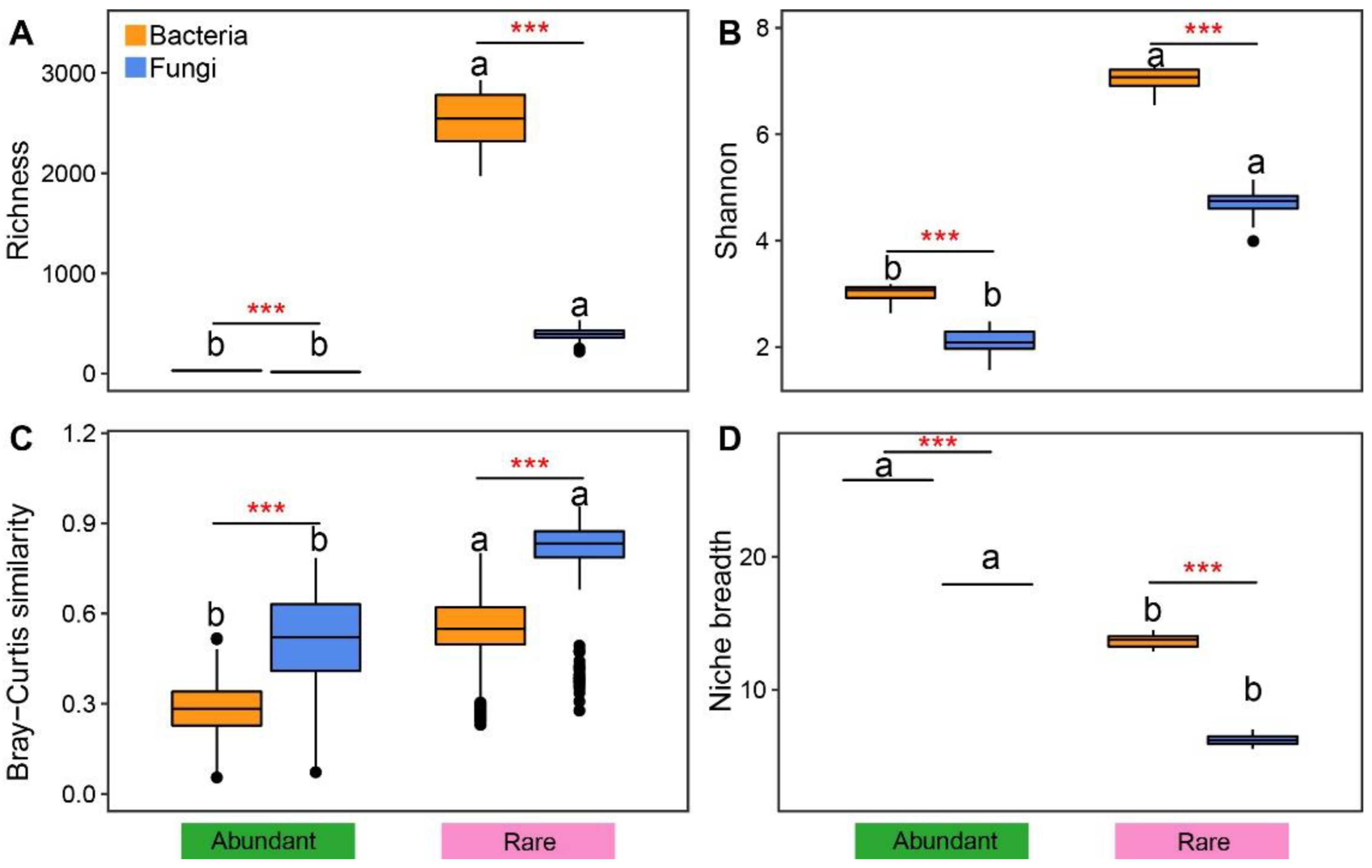
3.1. General Distribution of Rare and Abundant Taxa
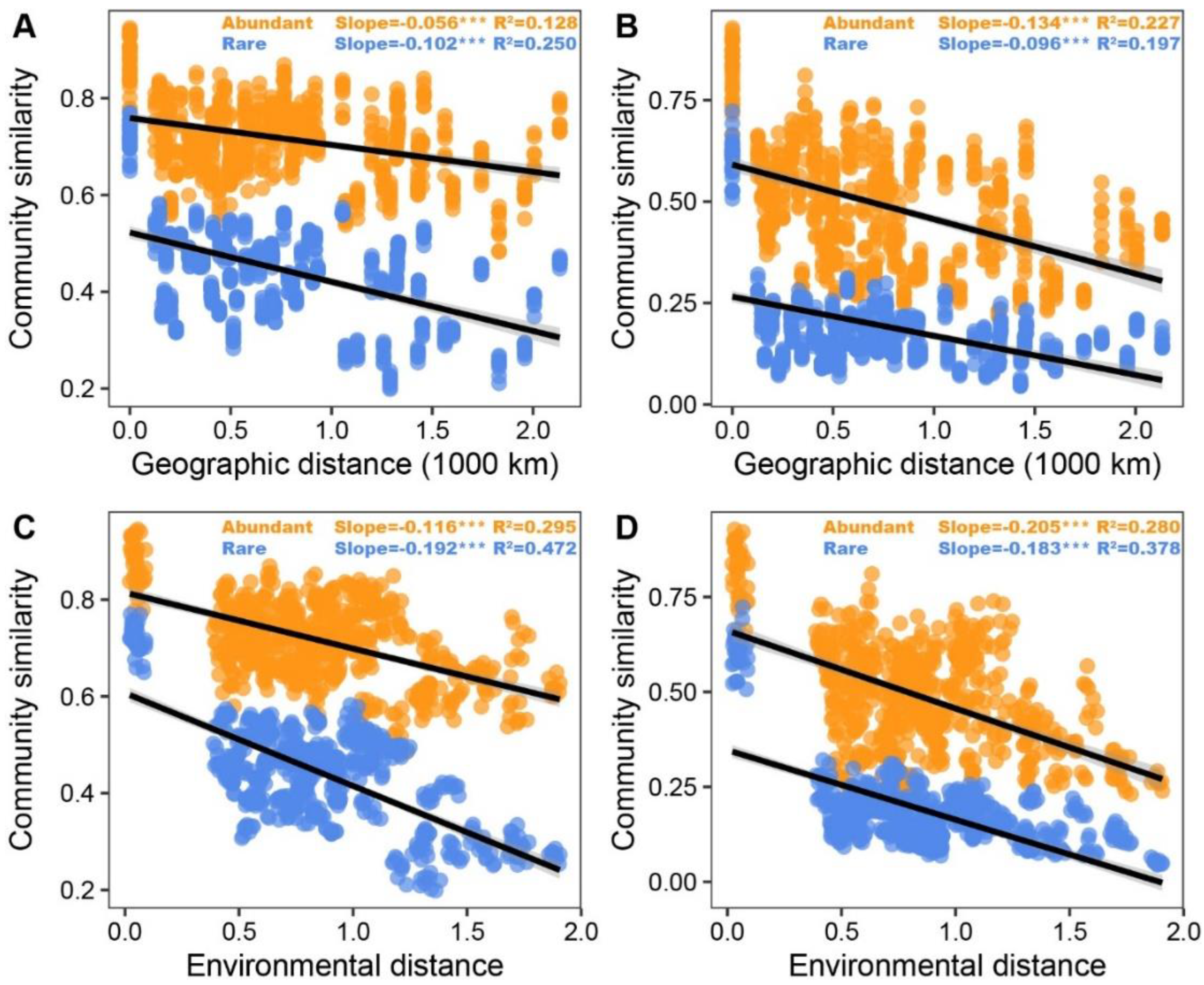
3.2. Patterns and Driving Factors of Beta-Diversity in Abundant and Rare Taxa
3.3. Assembly Processes in Abundant and Rare Taxa
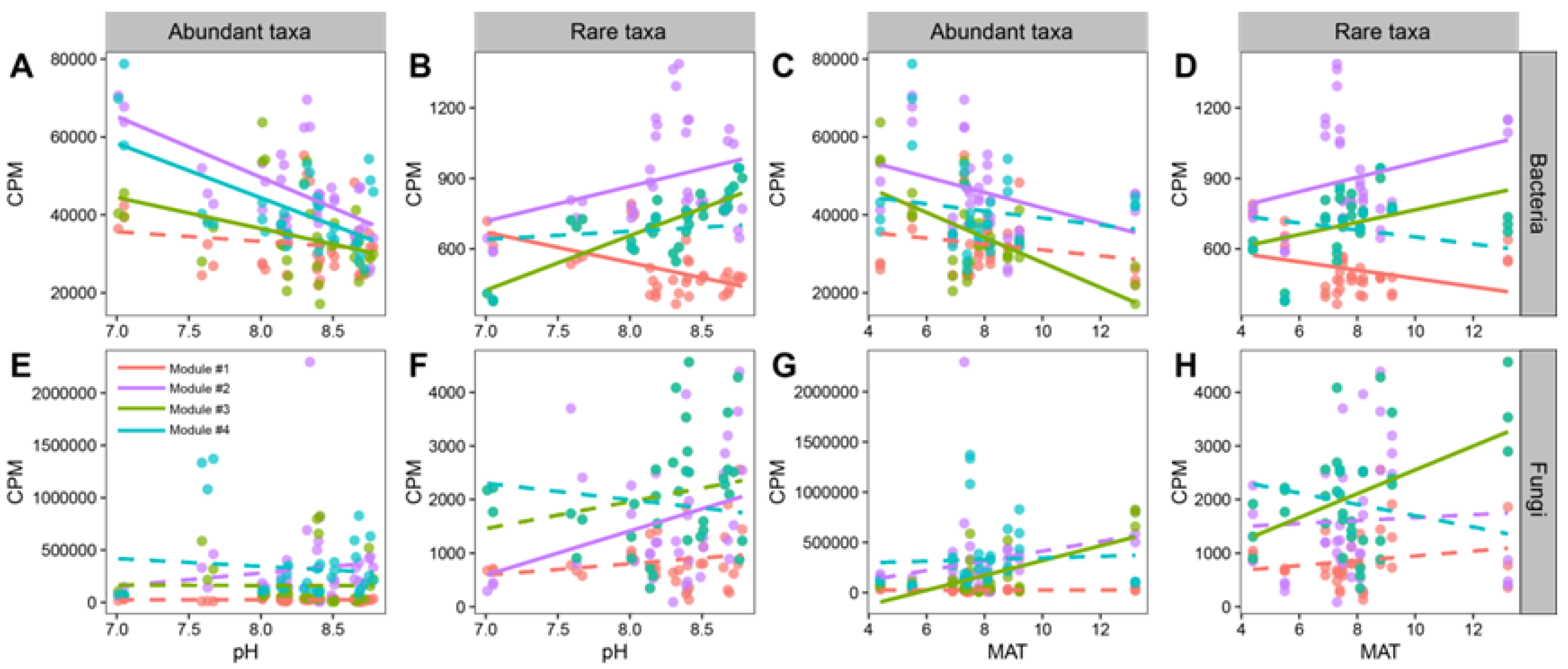
3.4. Co-Occurrence Patterns of Abundant and Rare Taxa
4. Discussion
4.1. Distribution Patterns of Abundant and Rare Microbes
4.2. Biogeographic Patterns of Abundant and Rare Microbes
4.3. Assembly Processes of Abundant and Rare Microbes
4.4. Co-Occurrence Patterns of Abundant and Rare Microbes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jiao, S.; Lu, Y. Abundant fungi adapt to broader environmental gradients than rare fungi in agricultural fields. Glob. Chang. Biol. 2020, 26, 4506–4520. [Google Scholar] [CrossRef]
- Madsen, E.L. Microorganisms and their roles in fundamental biogeochemical cycles. Curr. Opin. Biotechnol. 2011, 22, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Prosser, J.I.; Bohannan, B.J.M.; Curtis, T.P.; Ellis, R.J.; Firestone, M.K.; Freckleton, R.P.; Green, J.L.; Green, L.E.; Killham, K.; Lennon, J.J.; et al. The role of ecological theory in microbial ecology. Nat. Rev. Genet. 2007, 5, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, N.; Beßler, H.; Engels, C.; Gleixner, G.; Habekost, M.; Milcu, A.; Partsch, S.; Sabais, A.C.W.; Scherber, C.; Steinbeiss, S.; et al. Plant diversity effects on soil microorganisms support the singular hypothesis. Ecology 2010, 91, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Pedrós-Alió, C. The rare bacterial biosphere. Annu. Rev. Mar. Sci. 2012, 4, 449–466. [Google Scholar] [CrossRef]
- Dini-Andreote, F.; de Cássia Pereira e Silva, M.; Triadó-Margarit, X.; Casamayor, E.O.; Van Elsas, J.D.; Salles, J.F. Dynamics of bacterial community succession in a salt marsh chronosequence: Evidences for temporal niche partitioning. ISME J. 2014, 8, 1989–2001. [Google Scholar] [CrossRef]
- Elshahed, M.S.; Youssef, N.; Spain, A.M.; Sheik, C.; Najar, F.Z.; Sukharnikov, L.O.; Roe, B.A.; Davis, J.; Schloss, P.D.; Bailey, V.; et al. Novelty and Uniqueness Patterns of Rare Members of the Soil Biosphere. Appl. Environ. Microbiol. 2008, 74, 5422–5428. [Google Scholar] [CrossRef]
- Xue, Y.; Chen, H.; Yang, J.R.; Liu, M.; Huang, B. Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J. 2018, 12, 2263–2277. [Google Scholar] [CrossRef]
- Pester, M.; Bittner, N.; Deevong, P.; Wagner, M.; Loy, A. A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J. 2010, 4, 1591–1602. [Google Scholar] [CrossRef]
- Liao, J.; Cao, X.; Zhao, L.; Wang, J.; Gao, Z.; Wang, M.C.; Huang, Y. The importance of neutral and niche processes for bacterial community assembly differs between habitat generalists and specialists. FEMS Microbiol. Ecol. 2016, 92, fiw174. [Google Scholar] [CrossRef]
- Liu, L.; Yang, J.; Yu, Z.; Wilkinson, D.M. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. 2015, 9, 2068–2077. [Google Scholar] [CrossRef]
- He, Z.; Liu, D.; Shi, Y.; Wu, X.; Dai, Y.; Shang, Y.; Peng, J.; Cui, Z. Broader environmental adaptation of rare rather than abundant bacteria in reforestation succession soil. Sci. Total. Environ. 2022, 828, 154364. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Wang, H.; Dsouza, M.; Lou, J.; He, Y.; Dai, Z.; Brookes, P.C.; Xu, J.; Gilbert, J.A. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J. 2016, 10, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef] [PubMed]
- Eiler, A.; Heinrich, F.; Bertilsson, S. Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J. 2012, 6, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Dini-Andreote, F.; Zhang, N.; Liang, C.; Yao, Z.; Zhang, H.; Zhang, D. Divergent Co-occurrence Patterns and Assembly Processes Structure the Abundant and Rare Bacterial Communities in a Salt Marsh Ecosystem. Appl. Environ. Microbiol. 2020, 86, e00322-20. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.; Wei, G. Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils. Mol. Ecol. 2017, 26, 5305–5317. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, F.; Xie, W.; Wang, K.; Zhou, X.; Zhang, D.; Zhu, X. Interaction and assembly processes of abundant and rare microbial communities during a diatom bloom process. Environ. Microbiol. 2020, 22, 1707–1719. [Google Scholar] [CrossRef]
- Tian, L.; Wang, Y.; Yang, J.; Zhang, L.; Feng, B. Rhizosphere bacterial community structure of three minor grain crops: A case-study from paired field sites in northern China. Land Degrad. Dev. 2022, 33, 104–116. [Google Scholar] [CrossRef]
- Tian, L.; Chen, P.; Gao, Z.; Gao, X.; Feng, B. Deciphering the distinct mechanisms shaping the broomcorn millet rhizosphere bacterial and fungal communities in a typical agricultural ecosystem of Northern China. Plant Soil 2022, 474, 469–484. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Glckner, F.O. The silva ribosomal rna gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Mangot, J.-F.; Domaizon, I.; Taib, N.; Marouni, N.; Duffaud, E.; Bronner, G.; Debroas, D. Short-term dynamics of diversity patterns: Evidence of continual reassembly within lacustrine small eukaryotes. Environ. Microbiol. 2013, 15, 1745–1758. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Zhang, Y.; Tang, Y.; Bai, Y.; Tao, Y.; Huang, B.; Wen, D. Identifying the key taxonomic categories that characterize microbial community diversity using full-scale classification: A case study of microbial communities in the sediments of Hangzhou Bay. FEMS Microbiol. Ecol. 2016, 92, 1–11. [Google Scholar] [CrossRef]
- Levins, R. Evolution in Changing Environments; Princeton University Press: Princeton, NJ, USA, 1968. [Google Scholar]
- Zhang, J.; Zhang, M. Spaa: Species Association Analysis. R package Version 0.2.1. 2013. Available online: https://cran.r-project.org/package=spaa (accessed on 12 January 2022).
- Martiny, J.; Bohannan, B.J.M.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; et al. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Genet. 2006, 4, 102–112. [Google Scholar] [CrossRef]
- Naimi, B.; Hamm, N.A.S.; Groen, T.A.; Skidmore, A.K.; Toxopeus, A.G. Where is positional uncertainty important for species distribution modelling? Ecography 2014, 37, 191–203. [Google Scholar] [CrossRef]
- Ning, D.; Deng, Y.; Tiedje, J.M.; Zhou, J. A general framework for quantitatively assessing ecological stochasticity. Proc. Natl. Acad. Sci. USA 2019, 116, 16892–16898. [Google Scholar] [CrossRef]
- Ju, F.; Xia, Y.; Guo, F.; Wang, Z.; Zhang, T. Taxonomic relatedness shapes bacterial assembly in activated sludge of globally distributed wastewater treatment plants. Environ. Microbiol. 2014, 16, 2421–2432. [Google Scholar] [CrossRef]
- Harrell, F.J.; Dupont, C. Hmisc: Harrell Miscellaneous. R Package Version 4.0-3 Online Publication. 2017. Available online: https://hbiostat.org/R/Hmisc/ (accessed on 12 January 2022).
- Zheng, W.; Zhao, Z.; Lv, F.; Wang, R.; Wang, Z.; Zhao, Z.; Li, Z.; Zhai, B. Assembly of abundant and rare bacterial and fungal sub-communities in different soil aggregate sizes in an apple orchard treated with cover crop and fertilizer. Soil Biol. Biochem. 2021, 156, 108222. [Google Scholar] [CrossRef]
- Lynch, M.D.J.; Neufeld, J.D. Ecology and exploration of the rare biosphere. Nat. Rev. Genet. 2015, 13, 217–229. [Google Scholar] [CrossRef]
- Nolte, V.; Pandey, R.V.; Jost, S.; Medinger, R.; Ottenwälder, B.; Boenigk, J.; Schlötterer, C. Contrasting seasonal niche separation between rare and abundant taxa conceals the extent of protist diversity. Mol. Ecol. 2010, 19, 2908–2915. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Lu, Y. Soil pH and temperature regulate assembly processes of abundant and rare bacterial communities in agricultural ecosystems. Environ. Microbiol. 2020, 22, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Zhang, W.; Yang, J.; Lin, Y.; Yu, Z.; Lin, S. Biogeographic patterns of abundant and rare bacterioplankton in three subtropical bays resulting from selective and neutral processes. ISME J. 2018, 12, 2198–2210. [Google Scholar] [CrossRef]
- Logares, R.; Audic, S.; Bass, D.; Bittner, L.; Boutte, C.; Christen, R.; Claverie, J.-M.; Decelle, J.; Dolan, J.R.; Dunthorn, M.; et al. Patterns of Rare and Abundant Marine Microbial Eukaryotes. Curr. Biol. 2014, 24, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Ushio, M.; Fukui, S.; Kondoh, M. Functional diversity of microbial decomposers facilitates plant coexistence in a plant–microbe–soil feedback model. Proc. Natl. Acad. Sci. USA 2010, 107, 14251–14256. [Google Scholar] [CrossRef]
- Ji, M.; Kong, W.; Stegen, J.; Yue, L.; Wang, F.; Dong, X.; Cowan, D.A.; Ferrari, B.C. Distinct assembly mechanisms underlie similar biogeographical patterns of rare and abundant bacteria in Tibetan Plateau grassland soils. Environ. Microbiol. 2020, 22, 2261–2272. [Google Scholar] [CrossRef]
- Zhao, Z.; Ma, Y.; Feng, T.; Kong, X.; Wang, Z.; Zheng, W.; Zhai, B. Assembly processes of abundant and rare microbial communities in orchard soil under a cover crop at different periods. Geoderma 2022, 406, 115543. [Google Scholar] [CrossRef]
- Alonso-Sáez, L.; Díaz-Pérez, L.; Morán, X. The hidden seasonality of the rare biosphere in coastal marine bacterioplankton. Environ. Microbiol. 2015, 10, 3766–3780. [Google Scholar] [CrossRef]
- Astudillo-García, C.; Hermans, S.M.; Stevenson, B.; Buckley, H.L.; Lear, G. Microbial assemblages and bioindicators as proxies for ecosystem health status: Potential and limitations. Appl. Microbiol. Biotechnol. 2019, 103, 6407–6421. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Schmidt, S.K.; Fukami, T.; Neill, S.P.O.; Bilinski, T.M.; Stanish, L.F.; Knelman, J.E.; Darcy, J.L.; Lynch, R.C.; Wickey, P. Patterns and processes of microbial community assembly. Microbiol. Mol. Biol. Rev. 2013, 77, 342–356. [Google Scholar] [CrossRef]
- Wang, J.; Li, M.; Li, J. Soil pH and moisture govern the assembly processes of abundant and rare bacterial communities in a dryland montane forest. Environ. Microbiol. Rep. 2021, 13, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Gadd, G.M.; Yang, Y.; Yuan, W.; Gu, J.; Ye, L.; Liu, W. Environmental adaptation is stronger for abundant rather than rare microorganisms in wetland soils from the Qinghai-Tibet Plateau. Mol. Ecol. 2021, 30, 2390–2403. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Weisenhorn, P.; Gilbert, J.A.; Shi, Y.; Bai, Y.; Chu, H. Soil pH correlates with the co-occurrence and assemblage process of diazotrophic communities in rhizosphere and bulk soils of wheat fields. Soil Biol. Biochem. 2018, 121, 185–192. [Google Scholar] [CrossRef]
- Wang, J.; Shen, J.; Wu, Y.; Tu, C.; Soininen, J.; Stegen, J.; He, J.-Z.; Liu, X.; Zhang, L.; Zhang, E. Phylogenetic beta diversity in bacterial assemblages across ecosystems: Deterministic versus stochastic processes. ISME J. 2013, 7, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-B.; Lü, X.-T.; Yao, J.; Wang, Z.-W.; Deng, Y.; Cheng, W.-X.; Zhou, J.-Z.; Han, X.-G. Habitat-specific patterns and drivers of bacterial β-diversity in China’s drylands. ISME J. 2017, 11, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Deng, Y.; Wang, F.; Wang, W.; Xu, Z.; Hao, Y.; Xue, K.; Zhang, B.; Tang, L.; Zhou, H.; et al. Autotrophic and symbiotic diazotrophs dominate nitrogen-fixing communities in Tibetan grassland soils. Sci. Total. Environ. 2018, 639, 997–1006. [Google Scholar] [CrossRef]
- Wang, C.; Zheng, M.M.; Song, W.F.; Chen, R.F.; Zhao, X.Q.; Wen, S.L.; Zheng, Z.S.; Shen, R.F. Biogeographic patterns and co-occurrence networks of diazotrophic and arbuscular mycorrhizal fungal communities in the acidic soil ecosystem of southern China. Appl. Soil Ecol. 2021, 158, 103798. [Google Scholar] [CrossRef]
- Ostman, O.; Drakare, S.; Kritzberg, E.S.; Langenheder, S.; Logue, J.B.; Lindstrã¶m, E.S. Regional invariance among microbial communities. Ecol. Lett. 2010, 13, 118–127. [Google Scholar] [CrossRef]
- Wang, K.; Ye, X.; Chen, H.; Zhao, Q.; Hu, C.; He, J.; Qian, Y.; Xiong, J.; Zhu, J.; Zhang, D. Bacterial biogeography in the coastal waters of northern Zhejiang, East China Sea is highly controlled by spatially structured environmental gradients. Environ. Microbiol. 2015, 17, 3898–3913. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef]
- Hou, J.; Wu, L.; Liu, W.; Ge, Y.; Mu, T.; Zhou, T.; Li, Z.; Zhou, J.; Sun, X.; Luo, Y.; et al. Biogeography and diversity patterns of abundant and rare bacterial communities in rice paddy soils across China. Sci. Total. Environ. 2020, 730, 139116. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, Y.; Xiang, X.; Sun, R.; Yang, T.; He, D.; Zhang, K.; Ni, Y.; Zhu, Y.-G.; Adams, J.; et al. Spatial scale affects the relative role of stochasticity versus determinism in soil bacterial communities in wheat fields across the North China Plain. Microbiome 2018, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, E.M.; Mau, R.L.; Hayer, M.; Liu, X.-J.A.; Schwartz, E.; Dijkstra, P.; Koch, B.J.; Allen, K.; Blazewicz, S.J.; Hofmockel, K.; et al. Evolutionary history constrains microbial traits across environmental variation. Nat. Ecol. Evol. 2019, 3, 1064–1069. [Google Scholar] [CrossRef]
- Palomo, A.; Pedersen, A.G.; Fowler, S.J.; Dechesne, A.; Sicheritz-Pontén, T.; Smets, B.F. Comparative genomics sheds light on niche differentiation and the evolutionary history of comammox Nitrospira. ISME J. 2017, 12, 1779–1793. [Google Scholar] [CrossRef]
- Zinger, L.; Taberlet, P.; Schimann, H.; Bonin, A.; Boyer, F.; De Barba, M.; Gaucher, P.; Gielly, L.; Giguet-Covex, C.; Iribar, A.; et al. Body size determines soil community assembly in a tropical forest. Mol. Ecol. 2018, 28, 528–543. [Google Scholar] [CrossRef]
- Bailey, R.I.; Freerk, M.; Chloe, V.; Steffen, W.; Andreas, P. Large body size constrains dispersal assembly of communities even across short distances. Sci. Rep. 2018, 8, 10911. [Google Scholar] [PubMed]
- Gao, C.; Montoya, L.; Xu, L.; Madera, M.; Hollingsworth, J.; Purdom, E.; Singan, V.; Vogel, J.; Hutmacher, R.B.; Dahlberg, J.A.; et al. Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nat. Commun. 2020, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Xun, W.; Li, W.; Xiong, W.; Ren, Y.; Liu, Y.; Miao, Y.; Xu, Z.; Zhang, N.; Shen, Q.; Zhang, R. Diversity-triggered deterministic bacterial assembly constrains community functions. Nat. Commun. 2019, 10, 3833. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; He, Z.; Zhou, J.; Firestone, M.K. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef]
- Hu, A.; Ju, F.; Hou, L.; Li, J.; Yang, X.; Wang, H.; Mulla, S.I.; Sun, Q.; Bürgmann, H.; Yu, C.-P. Strong impact of anthropogenic contamination on the co-occurrence patterns of a riverine microbial community. Environ. Microbiol. 2017, 19, 4993–5009. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, F.; Wu, S.; Wu, J.; Yan, J.; Xu, K.; Hong, Y. Biogeographic pattern of bacterioplanktonic community and potential function in the Yangtze River: Roles of abundant and rare taxa. Sci. Total. Environ. 2020, 747, 141335. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.; Eguíluz, V.M.; Duarte, C.M.; Voolstra, C.R. Rare symbionts may contribute to the resilience of coral–algal assemblages. ISME J. 2018, 12, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Jousset, A.; Guo, S.; Karlsson, I.; Zhao, Q.; Wu, H.; Kowalchuk, G.A.; Shen, Q.; Li, R.; Geisen, S. Soil protist communities form a dynamic hub in the soil microbiome. ISME J. 2017, 12, 634–638. [Google Scholar] [CrossRef]
- Guimerà, R.; Amaral, L.A.N. Functional cartography of complex metabolic networks. Nature 2005, 433, 895–900. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, J.; Liu, Y.; Shi, P.; Wei, G. Co-occurrence patterns of soybean rhizosphere microbiome at a continental scale. Soil Biol. Biochem. 2018, 118, 178–186. [Google Scholar] [CrossRef]
- Jiao, S.; Xu, Y.; Zhang, J.; Hao, X.; Lu, Y. Core Microbiota in Agricultural Soils and Their Potential Associations with Nutrient Cycling. mSystems 2019, 4, e00313-18. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. Uchime improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abundant Taxa (AT) | Rare Taxa (RT) | Moderate Taxa (MT) | Conditionally Rare and Abundant Taxa (CRAT) | ||
|---|---|---|---|---|---|
| OTU number (%) | Bacteria | 30 (0.38%) | 8227 (97.28%) | 190 (2.25%) | 10 (0.12%) |
| Fungi | 17 (0.62%) | 2623 (95.84%) | 2 (0.07%) | 95 (3.47%) |
| Bacteria | Fungi | |||
|---|---|---|---|---|
| Abundant | Rare | Abundant | Rare | |
| pH | 0.432 *** | 0.525 *** | 0.392 *** | 0.431 *** |
| OM | 0.267 ** | 0.257 ** | 0.331 *** | 0.232 ** |
| TN | 0.278 *** | 0.258 ** | 0.203 ** | 0.234 *** |
| AP | 0.189 * | 0.125 | 0.105 | 0.177 * |
| AK | −0.064 | −0.050 | 0.060 | 0.013 |
| NH4+-N | 0.065 | 0.252 *** | −0.016 | 0.275 *** |
| NO3−-N | 0.026 | 0.048 | 0.229 ** | 0.158 * |
| MAT | 0.478 *** | 0.449 *** | 0.592 *** | 0.541 *** |
| MAP | 0.032 | 0.074 | 0.087 | 0.132 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, L.; Zhang, Y.; Zhang, L.; Zhang, L.; Gao, X.; Feng, B. Biogeographic Pattern and Network of Rhizosphere Fungal and Bacterial Communities in Panicum miliaceum Fields: Roles of Abundant and Rare Taxa. Microorganisms 2023, 11, 134. https://doi.org/10.3390/microorganisms11010134
Tian L, Zhang Y, Zhang L, Zhang L, Gao X, Feng B. Biogeographic Pattern and Network of Rhizosphere Fungal and Bacterial Communities in Panicum miliaceum Fields: Roles of Abundant and Rare Taxa. Microorganisms. 2023; 11(1):134. https://doi.org/10.3390/microorganisms11010134
Chicago/Turabian StyleTian, Lixin, Yuchuan Zhang, Liyuan Zhang, Lei Zhang, Xiaoli Gao, and Baili Feng. 2023. "Biogeographic Pattern and Network of Rhizosphere Fungal and Bacterial Communities in Panicum miliaceum Fields: Roles of Abundant and Rare Taxa" Microorganisms 11, no. 1: 134. https://doi.org/10.3390/microorganisms11010134
APA StyleTian, L., Zhang, Y., Zhang, L., Zhang, L., Gao, X., & Feng, B. (2023). Biogeographic Pattern and Network of Rhizosphere Fungal and Bacterial Communities in Panicum miliaceum Fields: Roles of Abundant and Rare Taxa. Microorganisms, 11(1), 134. https://doi.org/10.3390/microorganisms11010134