
Hybrid Genomic Analysis of Salmonella enterica Serovar Enteritidis SE3 Isolated from Polluted Soil in Brazil
 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Salmonella Isolation
2.2. DNA Isolation
2.3. Amplification of 16S rRNA Gene
2.4. 16S rRNA Gene Sequencing and Phylogenetic Analysis
2.5. Whole Genome Sequencing (WGS) by MinION and Illumina
2.6. Hybrid Genome Sequence Assembly
2.7. Serotype Identification
2.8. Gene Annotation
2.9. Genome Similarity Assessment
2.10. Pangenome Analysis
2.11. Mobile Genetic Element Identification and Annotation
3. Results
3.1. Salmonella Isolation and Characterization
3.2. Analysis of 16S rRNA
3.3. Whole Genome Sequencing of Salmonella Isolate SE3
3.4. Genome Assembly
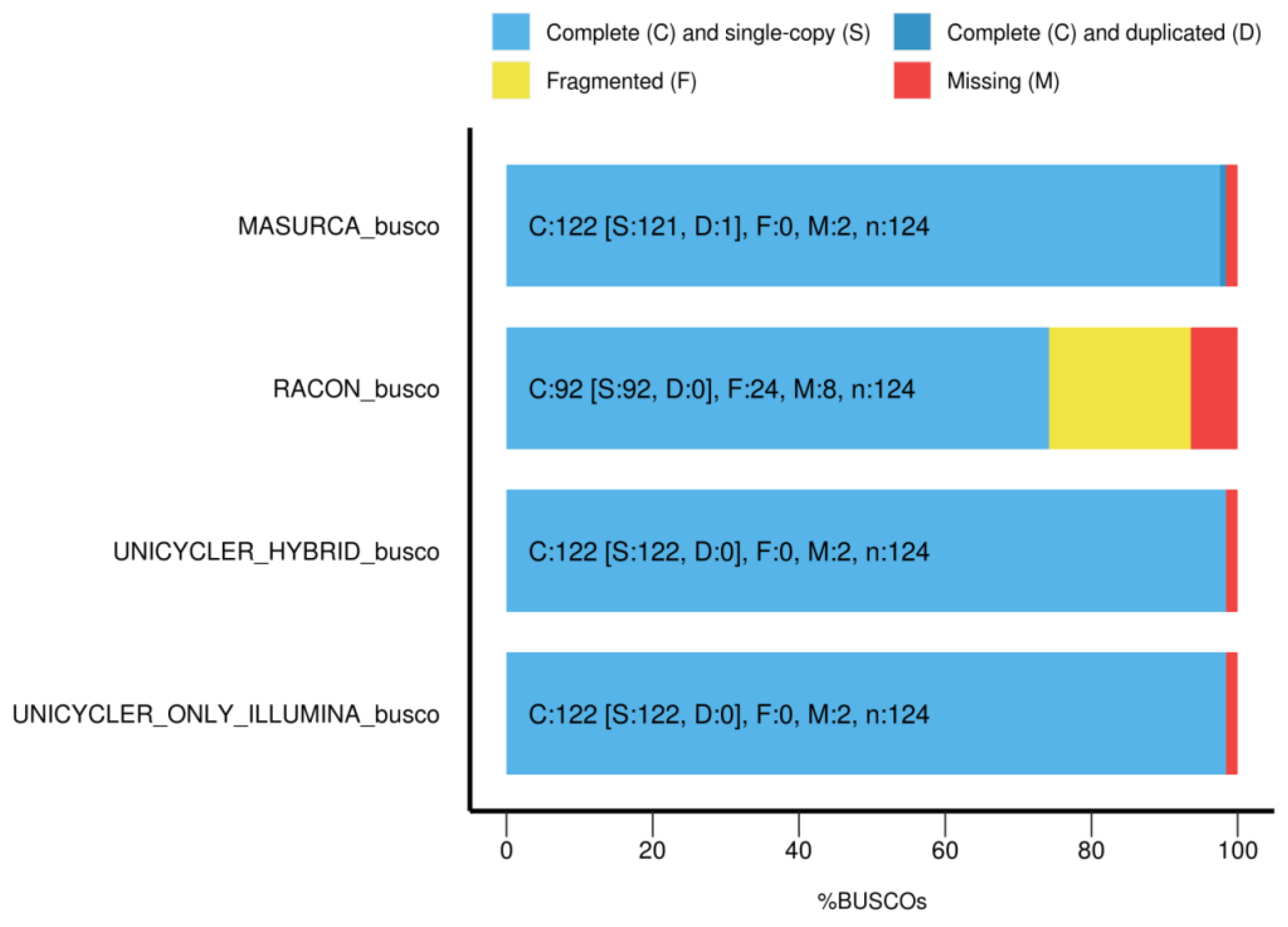
3.5. Completeness of the Genome Annotation
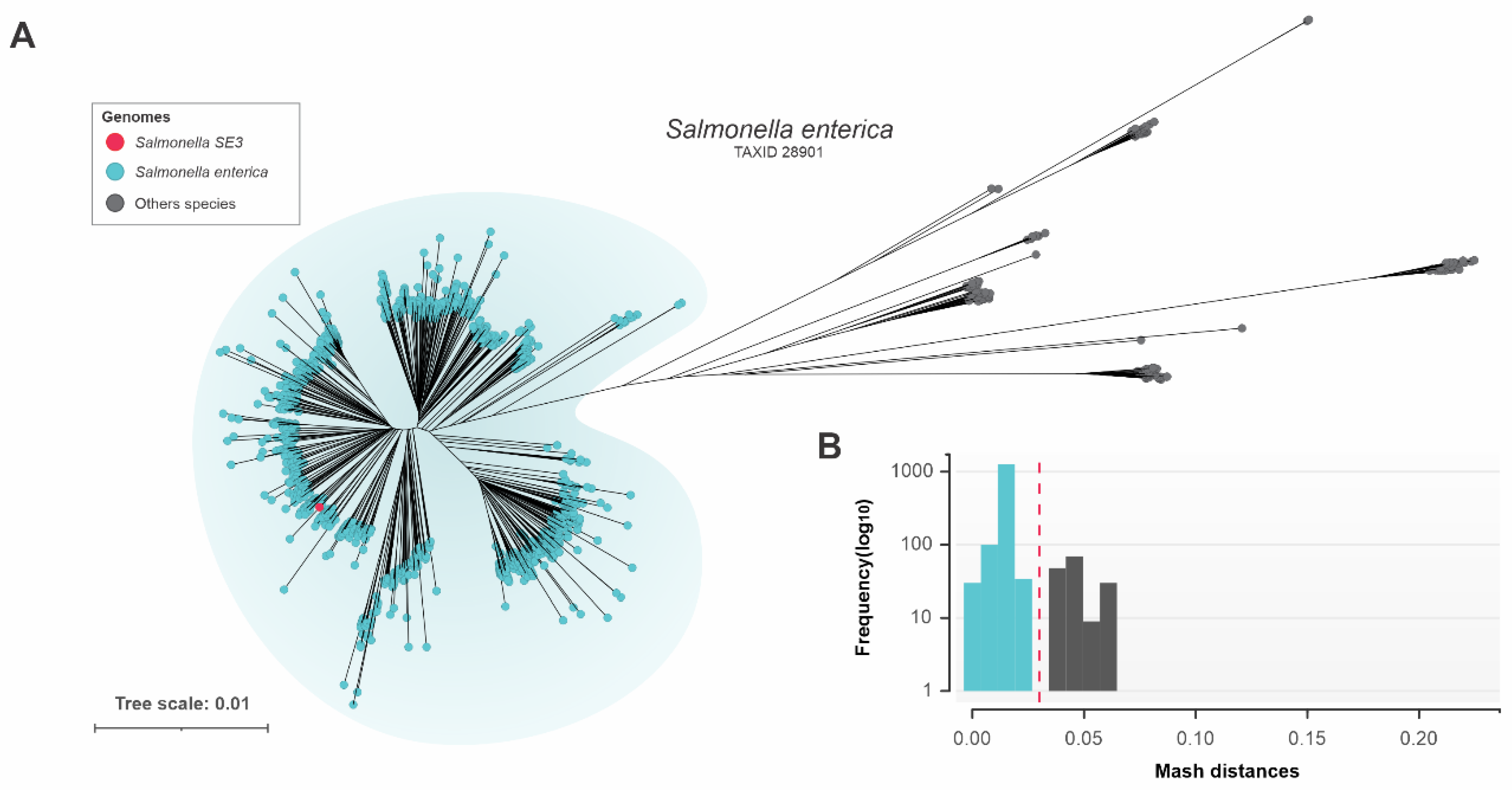
3.6. Genomic Relatedness of Salmonella SE3
3.7. Pangenome Analysis
3.8. Genome Features
3.8.1. Resistome Identification
3.8.2. Viriome, Genomic Island and Pathogenic Island Identification
3.9. Identification of Antiviral Defense Systems
3.10. Prophage Identification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hernández-Reyes, C.; Schikora, A. Salmonella, a Cross-Kingdom Pathogen Infecting Humans and Plants. FEMS Microbiol. Lett. 2013, 343, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Majowicz, S.E.; Musto, J.; Scallan, E.; Angulo, F.J.; Kirk, M.; O’Brien, S.J.; Jones, T.F.; Fazil, A.; Hoekstra, R.M.; International Collaboration on Enteric Disease “Burden of Illness” Studies. The Global Burden of Nontyphoidal Salmonella Gastroenteritis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2010, 50, 882–889. [Google Scholar] [CrossRef] [PubMed]
- The Global Burden of Non-Typhoidal Salmonella Invasive Disease: A Systematic Analysis for the Global Burden of Disease Study 2017—The Lancet Infectious Diseases. Available online: https://www.thelancet.com/journals/laninf/article/PIIS1473-3099(19)30418-9/fulltext (accessed on 2 November 2022).
- Das, S.; Ray, S.; Ryan, D.; Sahu, B.; Suar, M. Identification of a Novel Gene in ROD9 Island of Salmonella Enteritidis Involved in the Alteration of Virulence-Associated Genes Expression. Virulence 2018, 9, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.; Van Puyvelde, S.; Staes, A.; Timmerman, E.; Barbé, B.; Jacobs, J.; Gevaert, K.; Deborggraeve, S. Salmonella Typhi, Paratyphi A, Enteritidis and Typhimurium Core Proteomes Reveal Differentially Expressed Proteins Linked to the Cell Surface and Pathogenicity. PLoS Negl. Trop. Dis. 2019, 13, e0007416. [Google Scholar] [CrossRef]
- Rabsch, W.; Andrews, H.L.; Kingsley, R.A.; Prager, R.; Tschäpe, H.; Adams, L.G.; Bäumler, A.J. Salmonella enterica Serotype Typhimurium and Its Host-Adapted Variants. Infect. Immun. 2002, 70, 2249–2255. [Google Scholar] [CrossRef]
- Carden, S.; Okoro, C.; Dougan, G.; Monack, D. Non-Typhoidal Salmonella Typhimurium ST313 Isolates That Cause Bacte-remia in Humans Stimulate Less Inflammasome Activation than ST19 Isolates Associated with Gastroenteritis. Pathog. Dis. 2015, 73, ftu023. [Google Scholar] [CrossRef]
- Kipper, D.; Mascitti, A.K.; De Carli, S.; Carneiro, A.M.; Streck, A.F.; Fonseca, A.S.K.; Ikuta, N.; Lunge, V.R. Emergence, Dissemination and Antimicrobial Resistance of the Main Poultry-Associated Salmonella Serovars in Brazil. Vet. Sci. 2022, 9, 405. [Google Scholar] [CrossRef]
- Allard, M.W.; Strain, E.; Melka, D.; Bunning, K.; Musser, S.M.; Brown, E.W.; Timme, R. Practical Value of Food Pathogen Traceability through Building a Whole-Genome Sequencing Network and Database. J. Clin. Microbiol. 2016, 54, 1975–1983. [Google Scholar] [CrossRef]
- Gilchrist, C.A.; Turner, S.D.; Riley, M.F.; Petri, W.A.; Hewlett, E.L. Whole-Genome Sequencing in Outbreak Analysis. Clin. Microbiol. Rev. 2015, 28, 541–563. [Google Scholar] [CrossRef]
- Utturkar, S.M.; Klingeman, D.M.; Land, M.L.; Schadt, C.W.; Doktycz, M.J.; Pelletier, D.A.; Brown, S.D. Evaluation and Validation of de Novo and Hybrid Assembly Techniques to Derive High-Quality Genome Sequences. Bioinform. Oxf. Engl. 2014, 30, 2709–2716. [Google Scholar] [CrossRef]
- Ashton, P.M.; Nair, S.; Dallman, T.; Rubino, S.; Rabsch, W.; Mwaigwisya, S.; Wain, J.; O’Grady, J. MinION Nanopore Se-quencing Identifies the Position and Structure of a Bacterial Antibiotic Resistance Island. Nat. Biotechnol. 2015, 33, 296–300. [Google Scholar] [CrossRef]
- Genome Assembly Using Nanopore-Guided Long and Error-Free DNA Reads | BMC Genomics | Full Text. Available online: https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-015-1519-z (accessed on 2 November 2022).
- Genomic Analyses of Multidrug-Resistant Salmonella Indiana, Typhimurium, and Enteritidis Isolates Using MinION and MiSeq Sequencing Technologies | PLOS ONE. Available online: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0235641 (accessed on 2 November 2022).
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From Squiggle to Basepair: Computational Approaches for Improving Nanopore Sequencing Read Accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [PubMed]
- The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/27887629/ (accessed on 2 November 2022).
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. HybridSPAdes: An Algorithm for Hybrid Assembly of Short and Long Reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/28594827/ (accessed on 2 November 2022).
- Asai, T.; Otagiri, Y.; Osumi, T.; Namimatsu, T.; Hirai, H.; Sato, S. Isolation of Salmonella from Diarrheic Feces of Pigs. J. Vet. Med. Sci. 2002, 64, 159–160. [Google Scholar] [CrossRef]
- Senthilraj, R.; Prasad, G.S.; Janakiraman, K. Sequence-based identification of microbial contaminants in non-parenteral products. Braz. J. Pharm. 2016, 52, 329–336. [Google Scholar] [CrossRef]
- Molecular Evolution and Phylogenetics: Nei, Masatoshi, Kumar, Sudhir + Free Shipping. Available online: https://www.amazon.com/Molecular-Evolution-Phylogenetics-Masatoshi-Nei/dp/0195135857 (accessed on 2 November 2022).
- Tomé, L.M.R.; da Silva, F.F.; Fonseca, P.L.C.; Mendes-Pereira, T.; Azevedo, V.A.D.C.; Brenig, B.; Góes-Neto, A. Hybrid Assembly Improves Genome Quality and Completeness of Trametes villosa CCMB561 and Reveals a Huge Potential for Lignocellulose Breakdown. J. Fungi 2022, 8, 142. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive k-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and Accurate de Novo Genome Assembly from Long Uncorrected Reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- [PDF] Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM | Semantic Scholar. Available online: https://www.semanticscholar.org/paper/Aligning-sequence-reads%2C-clone-sequences-and-with-Li/74574ee09030e8aadb48fa349eb9b054e2f95ceb (accessed on 2 November 2022).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Hernandez, D.; François, P.; Farinelli, L.; Osterås, M.; Schrenzel, J. De Novo Bacterial Genome Sequencing: Millions of Very Short Reads Assembled on a Desktop Computer. Genome Res. 2008, 18, 802–809. [Google Scholar] [CrossRef]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA Genome Assembler. Bioinform. Oxf. Engl. 2013, 29, 2669–2677. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinform. Oxf. Engl. 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinform. Oxf. Engl. 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Fan, Y.; Wang, M.; Lv, J.; Zhang, H.; Sun, L.; Du, H. Effect of RpoE on the Non-Coding RNA Expression Profiles of Salmonella Enterica Serovar Typhi under the Stress of Ampicillin. Curr. Microbiol. 2020, 77, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinform. Oxf. Engl. 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast Genome and Metagenome Distance Estimation Using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries | Nature Communications. Available online: https://www.nature.com/articles/s41467-018-07641-9 (accessed on 2 November 2022).
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinform. Oxf. Engl. 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-Sites: Rapid Efficient Extraction of SNPs from Multi-FASTA Alignments. Microb. Genomics 2016, 2, e000056. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Interactive Tree of Life (ITOL) v4: Recent Updates and New Developments—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/30931475/ (accessed on 2 November 2022).
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded Prediction of Genomic Islands for Larger-Scale Datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A General Classification Scheme for Bacterial Virulence Factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- PADLOC: A Web Server for the Identification of Antiviral Defence Systems in Microbial Genomes | Nucleic Acids Research | Oxford Academic. Available online: https://academic.oup.com/nar/article/50/W1/W541/6593116?login=false (accessed on 2 November 2022).
- Tesson, F.; Hervé, A.; Mordret, E.; Touchon, M.; d’Humières, C.; Cury, J.; Bernheim, A. Systematic and Quantitative View of the Antiviral Arsenal of Prokaryotes. Nat. Commun. 2022, 13, 2561. [Google Scholar] [CrossRef]
- Roer, L.; Hendriksen, R.S.; Leekitcharoenphon, P.; Lukjancenko, O.; Kaas, R.S.; Hasman, H.; Aarestrup, F.M. Is the Evolution of Salmonella Enterica Subsp. Enterica Linked to Restriction-Modification Systems? mSystems 2016, 1, e00009-16. [Google Scholar] [CrossRef]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genomics 2011, 12, 402. [Google Scholar] [CrossRef]
- dos Santos, H.R.M.; Argolo, C.S.; Argôlo-Filho, R.C.; Loguercio, L.L. A 16S RDNA PCR-Based Theoretical to Actual Delta Approach on Culturable Mock Communities Revealed Severe Losses of Diversity Information. BMC Microbiol. 2019, 19, 74. [Google Scholar] [CrossRef] [PubMed]
- Vaid, R.K.; Thakur, Z.; Anand, T.; Kumar, S.; Tripathi, B.N. Comparative Genome Analysis of Salmonella enterica Serovar Gallinarum Biovars Pullorum and Gallinarum Decodes Strain Specific Genes. PLoS ONE 2021, 16, e0255612. [Google Scholar] [CrossRef] [PubMed]
- González-Escalona, N.; Allard, M.A.; Brown, E.W.; Sharma, S.; Hoffmann, M. Nanopore Sequencing for Fast Determination of Plasmids, Phages, Virulence Markers, and Antimicrobial Resistance Genes in Shiga Toxin-Producing Escherichia Coli. PloS ONE 2019, 14, e0220494. [Google Scholar] [CrossRef]
- Rapid, Multiplexed, Whole Genome and Plasmid Sequencing of Foodborne Pathogens Using Long-Read Nanopore Tech-nology | Scientific Reports. Available online: https://www.nature.com/articles/s41598-019-52424-x (accessed on 2 November 2022).
- Chand, Y.; Alam, M.A.; Singh, S. Pan-Genomic Analysis of the Species Salmonella enterica: Identification of Core Essential and Putative Essential Genes. Gene Rep. 2020, 20, 100669. [Google Scholar] [CrossRef]
- Park, S.-C.; Lee, K.; Kim, Y.O.; Won, S.; Chun, J. Large-Scale Genomics Reveals the Genetic Characteristics of Seven Species and Importance of Phylogenetic Distance for Estimating Pan-Genome Size. Front. Microbiol. 2019, 10, 834. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, F.A.; Brandelli, A.; Tondo, E.C. Antimicrobial Resistance in Salmonella Enteritidis from Foods Involved in Human Salmonellosis Outbreaks in Southern Brazil. New Microbiol. 2006, 29, 49–54. [Google Scholar] [PubMed]
- Vaz, C.S.L.; Streck, A.F.; Michael, G.B.; Marks, F.S.; Rodrigues, D.P.; Dos Reis, E.M.F.; Cardoso, M.R.I.; Canal, C.W. Anti-microbial Resistance and Subtyping of Salmonella enterica Subspecies Enterica Serovar Enteritidis Isolated from Human Outbreaks and Poultry in Southern Brazil. Poult. Sci. 2010, 89, 1530–1536. [Google Scholar] [CrossRef]
- Campioni, F.; Moratto Bergamini, A.M.; Falcão, J.P. Genetic Diversity, Virulence Genes and Antimicrobial Resistance of Salmonella Enteritidis Isolated from Food and Humans over a 24-Year Period in Brazil. Food Microbiol. 2012, 32, 254–264. [Google Scholar] [CrossRef]
- Achtman, M.; Wain, J.; Weill, F.-X.; Nair, S.; Zhou, Z.; Sangal, V.; Krauland, M.G.; Hale, J.L.; Harbottle, H.; Uesbeck, A.; et al. Multilocus Sequence Typing as a Replacement for Serotyping in Salmonella Enterica. PLoS Pathog. 2012, 8, e1002776. [Google Scholar] [CrossRef]
- Campioni, F.; Souza, R.A.; Martins, V.V.; Stehling, E.G.; Bergamini, A.M.M.; Falcão, J.P. Prevalence of GyrA Mutations in Nalidixic Acid-Resistant Strains of Salmonella Enteritidis Isolated from Humans, Food, Chickens, and the Farm Environment in Brazil. Microb. Drug Resist. Larchmt. N 2017, 23, 421–428. [Google Scholar] [CrossRef]
- Frye, J.; Jackson, C. Genetic Mechanisms of Antimicrobial Resistance Identified in Salmonella enterica, Escherichia coli, and Enteroccocus Spp. Isolated from U.S. Food Animals. Front. Microbiol. 2013, 4, 135. [Google Scholar]
- Mohakud, N.K.; Panda, R.K.; Patra, S.D.; Sahu, B.R.; Ghosh, M.; Kushwaha, G.S.; Misra, N.; Suar, M. Genome Analysis and Virulence Gene Expression Profile of a Multi Drug Resistant Salmonella enterica Serovar Typhimurium Ms202. Gut Pathog. 2022, 14, 28. [Google Scholar] [CrossRef]
- Vilela, F.P.; Rodrigues, D.D.P.; Ferreira, J.C.; Darini, A.L.D.C.; Allard, M.W.; Falcão, J.P. Genomic Characterization of Salmonella enterica Serovar Choleraesuis from Brazil Reveals a Swine Gallbladder Isolate Harboring Colistin Resistance Gene Mcr-1.1. Braz. J. Microbiol. Publ. Braz. Soc. Microbiol. 2022, 53, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Seribelli, A.A.; da Silva, P.; Frazão, M.R.; Kich, J.D.; Allard, M.W.; Falcão, J.P. Phylogenetic Relationship and Genomic Characterization of Salmonella Typhimurium Strains Isolated from Swine in Brazil. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2021, 93, 104977. [Google Scholar] [CrossRef]
- Borah, P.; Dutta, R.; Das, L.; Hazarika, G.; Choudhury, M.; Deka, N.K.; Malakar, D.; Hussain, M.I.; Barkalita, L.M. Prevalence, Antimicrobial Resistance and Virulence Genes of Salmonella Serovars Isolated from Humans and Animals. Vet. Res. Commun. 2022, 46, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Sharma, I. Detection of InvA Gene in Isolated Salmonella from Marketed Poultry Meat by PCR Assay. J. Food Process. Technol. 2016, 7, 2. [Google Scholar] [CrossRef]
- El-Sebay, N.A.; Shady, H.M.A.; El-Zeedy, S.A.E.-R.; Samy, A.A. InvA Gene Sequencing of Salmonella Typhimurium Isolated from Egyptian Poultry. Asian J. Sci. Res. 2017, 10, 194–202. [Google Scholar] [CrossRef][Green Version]
- Raffatellu, M.; Wilson, R.P.; Chessa, D.; Andrews-Polymenis, H.; Tran, Q.T.; Lawhon, S.; Khare, S.; Adams, L.G.; Bäumler, A.J. SipA, SopA, SopB, SopD, and SopE2 Contribute to Salmonella Enterica Serotype Typhimurium Invasion of Epithelial Cells. Infect. Immun. 2005, 73, 146–154. [Google Scholar] [CrossRef]
- Woodward, M.J.; Allen-Vercoe, E.; Redstone, J.S. Distribution, Gene Sequence and Expression in Vivo of the Plasmid Encoded Fimbrial Antigen of Salmonella Serotype Enteritidis. Epidemiol. Infect. 1996, 117, 17–28. [Google Scholar] [CrossRef]
- Nicholson, B.; Low, D. DNA Methylation-Dependent Regulation of Pef Expression in Salmonella Typhimurium. Mol. Microbiol. 2000, 35, 728–742. [Google Scholar] [CrossRef]
- Jackson, C.R.; Dugas, S.L. Phylogenetic Analysis of Bacterial and Archaeal ArsC Gene Sequences Suggests an Ancient, Common Origin for Arsenate Reductase. BMC Evol. Biol. 2003, 3, 18. [Google Scholar] [CrossRef]
- Pei, R.; Zhang, L.; Duan, C.; Gao, M.; Feng, R.; Jia, Q.; Huang, Z. (Jacky) Investigation of Stress Response Genes in Antimi-crobial Resistant Pathogens Sampled from Five Countries. Processes 2021, 9, 927. [Google Scholar] [CrossRef]
- Andrade, M.F.D.; Moraes, L.R.S. Lead Contamination in Santo Amaro Defies Decades of Research and Delayed Reaction on the Part of the Public Authorities. Ambiente Soc. 2013, 16, 63–80. [Google Scholar] [CrossRef]
- Carvalho, F.M.; Tavares, T.M.; Lins, L. Soil Contamination by a Lead Smelter in Brazil in the View of the Local Residents. Int. J. Environ. Res. Public Health 2018, 15, 2166. [Google Scholar] [CrossRef]
- Hardt, W.-D.; Urlaub, H.; Galán, J.E. A Substrate of the Centisome 63 Type III Protein Secretion System of Salmonella Typhimurium Is Encoded by a Cryptic Bacteriophage. Proc. Natl. Acad. Sci. USA 1998, 95, 2574–2579. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Bossi, N.; Uzzau, S.; Maloriol, D.; Bossi, L. Variable Assortment of Prophages Provides a Transferable Repertoire of Pathogenic Determinants in Salmonella. Mol. Microbiol. 2001, 39, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Switt, A.I.M.; Sulakvelidze, A.; Wiedmann, M.; Kropinski, A.M.; Wishart, D.S.; Poppe, C.; Liang, Y. Salmonella Phages and Prophages: Genomics, Taxonomy, and Applied Aspects. Methods Mol. Biol. Clifton NJ 2015, 1225, 237–287. [Google Scholar] [CrossRef]
- Worley, J.; Meng, J.; Allard, M.W.; Brown, E.W.; Timme, R.E. Salmonella enterica Phylogeny Based on Whole-Genome Se-quencing Reveals Two New Clades and Novel Patterns of Horizontally Acquired Genetic Elements. mBio 2018, 9, e02303-18. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Data | HiSeq | MinION |
|---|---|---|
| Reads | 15,997,283 | 13,326 |
| Total read bases (bp) | 7,999,481 | 67,978,671 |
| Mean coverage (%) | 51,185 | 13,590 |
| Longest read (bp) | 151 | 28,841 |
| Mean read length (bp) | 150 | 5101 |
| GC % | 52.00 | 52.18 |
| Genome size (bp) | 4,688,543 | 4,709,033 |
| Assembly Method | Racon | Unicycler | Edena | SPAdes | Unicycler | MaSuRCA |
|---|---|---|---|---|---|---|
| Sequence data | MinION | HiSeq | HiSeq | HiSeq | Hybrid | Hybrid |
| Number of contigs | 2 | 31 | 41 | 50 | 10 | 39 |
| Number of contigs (≥0 bp) | 2 | 65 | 54 | 111 | 18 | 42 |
| Number of contigs (≥50 kb) | 2 | 15 | 4,475,114 | 4,566,140 | 4 | 24 |
| Largest contigs | 4,671,311 | 1,262,086 | 488,615 | 1,276,166 | 2,750,500 | 519,108 |
| Total length (≥50 kb) | 4,730,597 | 4,683,367 | 4,701,851 | 4,805,245 | 4,713,463 | 4,585,719 |
| GC (%) | 52.18 | 52.14 | 52.15 | 51.85 | 52.16 | 52.15 |
| N50 | 4,671,311 | 478,501 | 181,604 | 491,607 | 2,750,500 | 246,991 |
| L50 | 1 | 3 | 8 | 3 | 1 | 7 |
| Assembly Method | Sequence Data | Complete (%) | Single Copy (%) | Duplicated (%) | Fragmented (%) | Missing (%) |
|---|---|---|---|---|---|---|
| Racon | MinION | 74.2 | 74.2 | 0 | 19.4 | 6.4 |
| Unicycler | HiSeq | 98.4 | 98.4 | 0 | 0 | 1.64 |
| Unicycler | Hybrid | 98.4 | 98.4 | 0 | 0 | 1.64 |
| MaSuRCA | Hybrid | 98.4 | 97.6 | 0.8 | 0 | 1.64 |
| Annotated Genome | Features |
|---|---|
| rRNA | 20 |
| tRNA | 87 |
| Repeat region | 2 |
| CDS | 4403 |
| mRNA | 1 |
| No | SPI | Identity | Query/Template Length | Salmonella Serotype | Insertion Site | Accession Number |
|---|---|---|---|---|---|---|
| 1 | SPI-1 | 99.7 | 2705/2705 | Typhimurium SL1344 | fhlA/mutS | AF148689 |
| 2 | SPI-2 | 100 | 642/642 | Gallinarum SGC_2 | tRNA-valV | AY956827 |
| 3 | SPI-3 | 99.05 | 738/738 | Typhimurium 14028s | tRNA-selC | AJ000509 |
| 4 | SPI-5 | 99.11 | 9069/9069 | Typhimurium LT2 | tRNA-serT | NC_003197 |
| 5 | SPI-10 | 98.28 | 553/554 | Gallinarum SGE_3 | Unpublished | AY956839 |
| 6 | SPI-11 | 98.54 | 9085/15686 | Choleraesuis SC_B67 | Gifsy-1 | NC_006905 |
| 7 | SPI-12 | 97.14 | 5766/11075 | Choleraesuis SC_B67 | tRNA-pro | NC_006905 |
| 8 | SPI-13 | 100 | 341/341 | Gallinarum SGA_10 | tRNA-pheV | AY956834 |
| 9 | SPI-14 | 99.8 | 501/501 | Gallinarum SGA_8 | Unpublished | AY956835 |
| 10 | C63PI | 99.12 | 4000/4000 | Typhimurium SL1344 | fhlA | AF128999 |
| 11 | CS54 | 98.09 | 19669/25252 | Typhimurium ATCC_14028 | xseA-yfgK | AF140550 |
| 12 | Unnamed | 100 | 330/330 | Enteritidis CMCC50041 | -- | JQ071613 |
| Number | System | Subtype | Tool | Reference |
|---|---|---|---|---|
| 1 | AbiU | AbiU | PADLOC | [46] |
| 2 | Cas type IE | Cas3e | PADLOC | [46] |
| 3 | Cas type IE | Cas8e | PADLOC | [46] |
| 4 | Cas type IE | Cas11e | PADLOC | [46] |
| 5 | Cas type IE | Cas7e | PADLOC | [46] |
| 6 | Cas type IE | Cas5e | PADLOC | [46] |
| 7 | Cas type IE | Cas6e | PADLOC | [46] |
| 8 | Cas type IE | Cas1e | PADLOC | [46] |
| 9 | Cas type IE | Cas2e | PADLOC | [46] |
| 10 | CBASS_type_I | Cyclase | PADLOC | [46] |
| 11 | CBASS_type_I | Effector | PADLOC | [46] |
| 12 | CRISPR array | CRISPR array | PADLOC | [46] |
| 13 | CRISPR array | CRISPR array | PADLOC | [46] |
| 14 | RM type I | Mtase I | PADLOC | [46] |
| 15 | RM type I | Specificity I | PADLOC | [46] |
| 16 | RM type I | Rease I | PADLOC | [46] |
| 17 | RM type II | Rease II | PADLOC | [46] |
| 18 | RM type II | Mtase II | PADLOC | [46] |
| 19 | RM type III | Rease III | PADLOC | [46] |
| 20 | RM type III | Mtase III | PADLOC | [46] |
| 21 | Cas Class1 subtype I E1 | Cas3 I 5 | DefenseFinder | [47] |
| 22 | Cas Class1 subtype I E1 | Cas8e I E 1 | DefenseFinder | [47] |
| 23 | Cas Class1 subtype I E1 | Cas2gr11 I E 2 | DefenseFinder | [47] |
| 24 | Cas Class1 subtype I E1 | Cas7 I E 2 | DefenseFinder | [47] |
| 25 | Cas Class1 subtype I E1 | Cas5 I E 3 | DefenseFinder | [47] |
| 26 | Cas Class1 subtype I E1 | Cas6e I II II IV V VI 1 | DefenseFinder | [47] |
| 27 | Cas Class1 subtype I E1 | Cas 1 I E 1 | DefenseFinder | [47] |
| 28 | Cas Class1 subtype I E1 | Cas2 I E 2 | DefenseFinder | [47] |
| 29 | CBASS I 2 | Cyclase SMODS | DefenseFinder | [47] |
| 30 | CBASS I 2 | 2TM Gros | DefenseFinder | [47] |
| 31 | RM Type III 2 | Type III Reases | DefenseFinder | [47] |
| 32 | RM Type III 2 | Type III Mtases | DefenseFinder | [47] |
| 33 | RM Type I 1 | Type I S | DefenseFinder | [47] |
| 34 | RM Type I 1 | Type I Mtases | DefenseFinder | [47] |
| 35 | RM Type I 1 | Type I S | DefenseFinder | [47] |
| 36 | RM Type I 1 | Type I Reases | DefenseFinder | [47] |
| Completeness | Score | Proteins | Position | Best Match | Accession No. | GC (%) |
|---|---|---|---|---|---|---|
| Incomplete | 60 | 27 | 805989–831780 | Shigella phage POCJ13 | NC_025434 | 48.7 |
| Intact | 150 | 40 | 1041034–1072153 | Salmonella phage Gifsy-2 | NC_010393 | 47.2 |
| Incomplete | 50 | 13 | 1276587–1286489 | Salmonella phage Gifsy-2 | NC_010393 | 46.7 |
| Incomplete | 30 | 9 | 1698977–1705339 | Shigella phage POCJ13 | NC_025434 | 45.6 |
| Intact | 150 | 49 | 1081056–1124788 | Salmonella phage RE-2010 | NC_019488 | 51.2 |
| Incomplete | 20 | 8 | 1435195–1442595 | Escherichia phage 500465-2 | NC_049343 | 53.2 |
| Incomplete | 40 | 9 | 29216–37324 | Salmonella phage RE-2010 | NC_019488 | 52.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-Calle, D.X.; Pedrosa-Silva, F.; Tomé, L.M.R.; Sousa, T.J.; de Oliveira Santos, L.T.S.; de Carvalho Azevedo, V.A.; Brenig, B.; Benevides, R.G.; Venancio, T.M.; Billington, C.; et al. Hybrid Genomic Analysis of Salmonella enterica Serovar Enteritidis SE3 Isolated from Polluted Soil in Brazil. Microorganisms 2023, 11, 111. https://doi.org/10.3390/microorganisms11010111
Romero-Calle DX, Pedrosa-Silva F, Tomé LMR, Sousa TJ, de Oliveira Santos LTS, de Carvalho Azevedo VA, Brenig B, Benevides RG, Venancio TM, Billington C, et al. Hybrid Genomic Analysis of Salmonella enterica Serovar Enteritidis SE3 Isolated from Polluted Soil in Brazil. Microorganisms. 2023; 11(1):111. https://doi.org/10.3390/microorganisms11010111
Chicago/Turabian StyleRomero-Calle, Danitza Xiomara, Francisnei Pedrosa-Silva, Luiz Marcelo Ribeiro Tomé, Thiago J. Sousa, Leila Thaise Santana de Oliveira Santos, Vasco Ariston de Carvalho Azevedo, Bertram Brenig, Raquel Guimarães Benevides, Thiago M. Venancio, Craig Billington, and et al. 2023. "Hybrid Genomic Analysis of Salmonella enterica Serovar Enteritidis SE3 Isolated from Polluted Soil in Brazil" Microorganisms 11, no. 1: 111. https://doi.org/10.3390/microorganisms11010111
APA StyleRomero-Calle, D. X., Pedrosa-Silva, F., Tomé, L. M. R., Sousa, T. J., de Oliveira Santos, L. T. S., de Carvalho Azevedo, V. A., Brenig, B., Benevides, R. G., Venancio, T. M., Billington, C., & Góes-Neto, A. (2023). Hybrid Genomic Analysis of Salmonella enterica Serovar Enteritidis SE3 Isolated from Polluted Soil in Brazil. Microorganisms, 11(1), 111. https://doi.org/10.3390/microorganisms11010111