
Phylogenomic Placement of American Southwest-Associated Clinical and Veterinary Isolates Expands Evidence for Distinct Cryptococcus gattii VGVI

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global Burden of Disease of HIV-Associated Cryptococcal Meningitis: An Updated Analysis. Lancet Infect. Dis. 2017, 17, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Cuomo, C.A.; Rhodes, J.; Desjardins, C.A. Advances in Cryptococcus Genomics: Insights into the Evolution of Pathogenesis. Mem. Inst. Oswaldo Cruz 2018, 113, e170473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagen, F.; Khayhan, K.; Theelen, B.; Kolecka, A.; Polacheck, I.; Sionov, E.; Falk, R.; Parnmen, S.; Lumbsch, H.T.; Boekhout, T. Recognition of Seven Species in the Cryptococcus gattii/Cryptococcus neoformans Species Complex. Fungal. Genet. Biol. 2015, 78, 16–48. [Google Scholar] [CrossRef] [Green Version]
- Firacative, C.; Roe, C.C.; Malik, R.; Ferreira-Paim, K.; Escandón, P.; Sykes, J.E.; Castañón-Olivares, L.R.; Contreras-Peres, C.; Samayoa, B.; Sorrell, T.C.; et al. MLST and Whole-Genome-Based Population Analysis of Cryptococcus gattii VGIII Links Clinical, Veterinary and Environmental Strains, and Reveals Divergent Serotype Specific Sub-Populations and Distant Ancestors. PLOS Negl. Trop. Dis. 2016, 10, e0004861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyazika, T.K.; Hagen, F.; Meis, J.F.; Robertson, V.J. Cryptococcus tetragattii as a Major Cause of Cryptococcal Meningitis among HIV-Infected Individuals in Harare, Zimbabwe. J. Infect. 2016, 72, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Farrer, R.A.; Chang, M.; Davis, M.J.; van Dorp, L.; Yang, D.-H.; Shea, T.; Sewell, T.R.; Meyer, W.; Balloux, F.; Edwards, H.M.; et al. A New Lineage of Cryptococcus gattii (VGV) Discovered in the Central Zambezian Miombo Woodlands. MBio 2019, 10, e02306-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, W.; Castañeda, A.; Jackson, S.; Huynh, M.; Castañeda, E. Molecular Typing of IberoAmerican Cryptococcus neoformans Isolates. Emerg. Infect. Dis. 2003, 9, 189–195. [Google Scholar] [CrossRef]
- Berejnoi, A.; Taverna, C.G.; Mazza, M.; Vivot, M.; Isla, G.; Córdoba, S.; Davel, G. First Case Report of Cryptococcosis Due to Cryptococcus decagattii in a Pediatric Patient in Argentina. Rev. Soc. Bras. Med. Trop. 2019, 52, e20180419. [Google Scholar] [CrossRef]
- Hagen, F.; Colom, M.F.; Swinne, D.; Tintelnot, K.; Iatta, R.; Montagna, M.T.; Torres-Rodriguez, J.M.; Cogliati, M.; Velegraki, A.; Burggraaf, A.; et al. Autochthonous and Dormant Cryptococcus gattii Infections in Europe. Emerg. Infect. Dis. 2012, 18, 1618–1624. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahl, J.W.; Lemmer, D.; Travis, J.; Schupp, J.M.; Gillece, J.D.; Aziz, M.; Driebe, E.M.; Drees, K.P.; Hicks, N.D.; Williamson, C.H.D.; et al. NASP: An Accurate, Rapid Method for the Identification of SNPs in WGS Datasets That Supports Flexible Input and Output Formats. Microb. Genom. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Schliep, K.P. Phangorn: Phylogenetic Analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, K.C. The Parsimony Ratchet, a New Method for Rapid Parsimony Analysis. Cladistics 1999, 15, 407–414. [Google Scholar] [CrossRef]
- Yu, G. Using Ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Casadevall, A.; Kontoyiannis, D.P.; Robert, V. On the Emergence of Candida Auris: Climate Change, Azoles, Swamps, and Birds. MBio 2019, 10, e01397-19. [Google Scholar] [CrossRef] [Green Version]
- Byrnes, E.J.; Li, W.; Lewit, Y.; Ma, H.; Voelz, K.; Ren, P.; Carter, D.A.; Chaturvedi, V.; Bildfell, R.J.; May, R.C.; et al. Emergence and Pathogenicity of Highly Virulent Cryptococcus gattii Genotypes in the Northwest United States. PLoS Pathog. 2010, 6, e1000850. [Google Scholar] [CrossRef] [Green Version]
- Cantrell, S.A.; Dianese, J.C.; Fell, J.; Gunde-Cimerman, N.; Zalar, P. Unusual Fungal Niches. Mycologia 2011, 103, 1161–1174. [Google Scholar] [CrossRef]
- Rosentreter, R.; Root, H.T. Biological Soil Crust Diversity and Composition in Southwest Idaho, USA. Bryologist 2019, 122, 10. [Google Scholar] [CrossRef]
- Warren, S.D.; St. Clair, L.L.; Johansen, J.R.; Kugrens, P.; Baggett, L.S.; Bird, B.J. Biological Soil Crust Response to Late Season Prescribed Fire in a Great Basin Juniper Woodland. Rangel. Ecol. Manag. 2015, 68, 241–247. [Google Scholar] [CrossRef]
- Mead, H.L.; Hamm, P.S.; Shaffer, I.N.; de Teixeira, M.M.; Wendel, C.S.; Wiederhold, N.P.; Thompson, G.R.; Muñiz-Salazar, R.; Castañón-Olivares, L.R.; Keim, P.; et al. Differential Thermotolerance Adaptation between Species of Coccidioides. J. Fungi 2020, 6, 366. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.; Smith, C.E.; Pappagianis, D.; Berman, R.J. Survival of Coccidioides Immitis Under Controlled Conditions of Temperature and Humidity. Am. J. Public Health Nations Health 1956, 46, 1317–1324. [Google Scholar] [CrossRef]
- Gorris, M.E.; Cat, L.A.; Zender, C.S.; Treseder, K.K.; Randerson, J.T. Coccidioidomycosis Dynamics in Relation to Climate in the Southwestern United States. Geohealth 2018, 2, 6–24. [Google Scholar] [CrossRef]
- Kollath, D.R.; Miller, K.J.; Barker, B.M. The Mysterious Desert Dwellers: Coccidioides immitis and Coccidioides posadasii, Causative Fungal Agents of Coccidioidomycosis. Virulence 2019, 10, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.W.; Barker, B.M. The Endozoan, Small-Mammal Reservoir Hypothesis and the Life Cycle of Coccidioides Species. Med. Mycol. 2019, 57, S16–S20. [Google Scholar] [CrossRef]
- del Rocío Reyes-Montes, M.; Pérez-Huitrón, M.A.; Ocaña-Monroy, J.L.; Frías-De-León, M.G.; Martínez-Herrera, E.; Arenas, R.; Duarte-Escalante, E. The Habitat of Coccidioides spp. and the Role of Animals as Reservoirs and Disseminators in Nature. BMC Infect. Dis. 2016, 16, 550. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues de Miranda, L. A New Genus: Sporopachydermia. Antonie Van Leeuwenhoek 1978, 44, 439–450. [Google Scholar] [CrossRef]
- Phaff, H.J.; Miller, M.W.; Miranda, M.; Heed, W.B.; Starmer, W.T. Original Papers Relating to the Systematics of Yeasts: Cryptococcus cereanus, a New Species of the Genus Cryptococcus. Int. J. Syst. Bacteriol. 1974, 24, 486–490. [Google Scholar] [CrossRef]
- Kingston, C.; Medinger, M.; Banderet-Uglioni, F.; Bassetti, S.; Bargetzi, M.; Haubitz, S.; Fux, C.A.; Bättig, V.; Goldenberger, D.; Passweg, J.; et al. Fungemia and Necrotic Lymph Node Infection with Sporopachydermia cereana in a Patient with Acute Myeloid Leukemia. Int. J. Infect. Dis. 2017, 61, 103–106. [Google Scholar] [CrossRef] [PubMed]
- al Dallal, H.A.; Narayanan, S.; Jones, C.M.; Lockhart, S.R.; Snyder, J.W. First Case Report of an Unusual Fungus (Sporopachydermia lactativora) Associated with a Pulmonary Infection in a Drug Injection User. Clin. Pathol. 2021, 14, 2632010X2110299. [Google Scholar] [CrossRef]
- Springer, D.J.; Billmyre, R.B.; Filler, E.E.; Voelz, K.; Pursall, R.; Mieczkowski, P.A.; Larsen, R.A.; Dietrich, F.S.; May, R.C.; Filler, S.G.; et al. Cryptococcus Gattii VGIII Isolates Causing Infections in HIV/AIDS Patients in Southern California: Identification of the Local Environmental Source as Arboreal. PLoS Pathog. 2014, 10, e1004285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firacative, C.; Trilles, L.; Meyer, W. MALDI-TOF MS Enables the Rapid Identification of the Major Molecular Types within the Cryptococcus neoformans/C. gattii Species Complex. PLoS ONE 2012, 7, e37566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posteraro, B.; Vella, A.; Cogliati, M.; de Carolis, E.; Florio, A.R.; Posteraro, P.; Sanguinetti, M.; Tortorano, A.M. Matrix-Assisted Laser Desorption Ionization–Time of Flight Mass Spectrometry-Based Method for Discrimination between Molecular Types of Cryptococcus neoformans and Cryptococcus gattii. J. Clin. Microbiol. 2012, 50, 2472–2476. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Chakrabarti, A. On the Origin of Candida Auris: Ancestor, Environmental Stresses, and Antiseptics. MBio 2020, 11, e02102-20. [Google Scholar] [CrossRef]
- Yadav, A.; Jain, K.; Wang, Y.; Pawar, K.; Kaur, H.; Sharma, K.K.; Tripathy, V.; Singh, A.; Xu, J.; Chowdhary, A. Candida auris on Apples: Diversity and Clinical Significance. MBio 2022, 13, e00518-22. [Google Scholar] [CrossRef]
- Casadevall, A.; Kontoyiannis, D.P.; Robert, V. Environmental Candida auris and the Global Warming Emergence Hypothesis. MBio 2021, 12, e00360-21. [Google Scholar] [CrossRef]
- Engelthaler, D.M.; Casadevall, A. On the Emergence of Cryptococcus gattii in the Pacific Northwest: Ballast Tanks, Tsunamis, and Black Swans. MBio 2019, 10, e02193-19. [Google Scholar] [CrossRef] [Green Version]
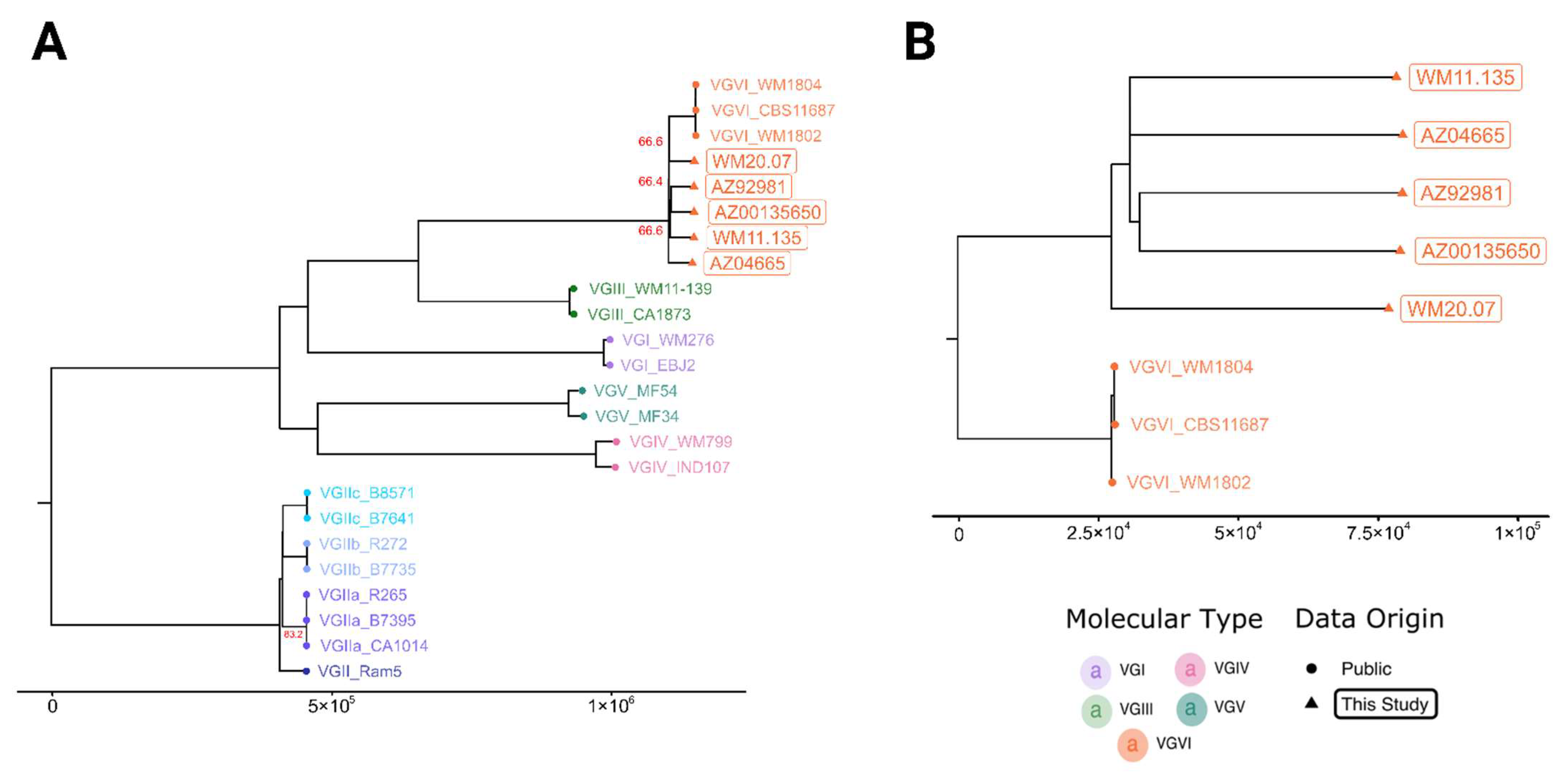

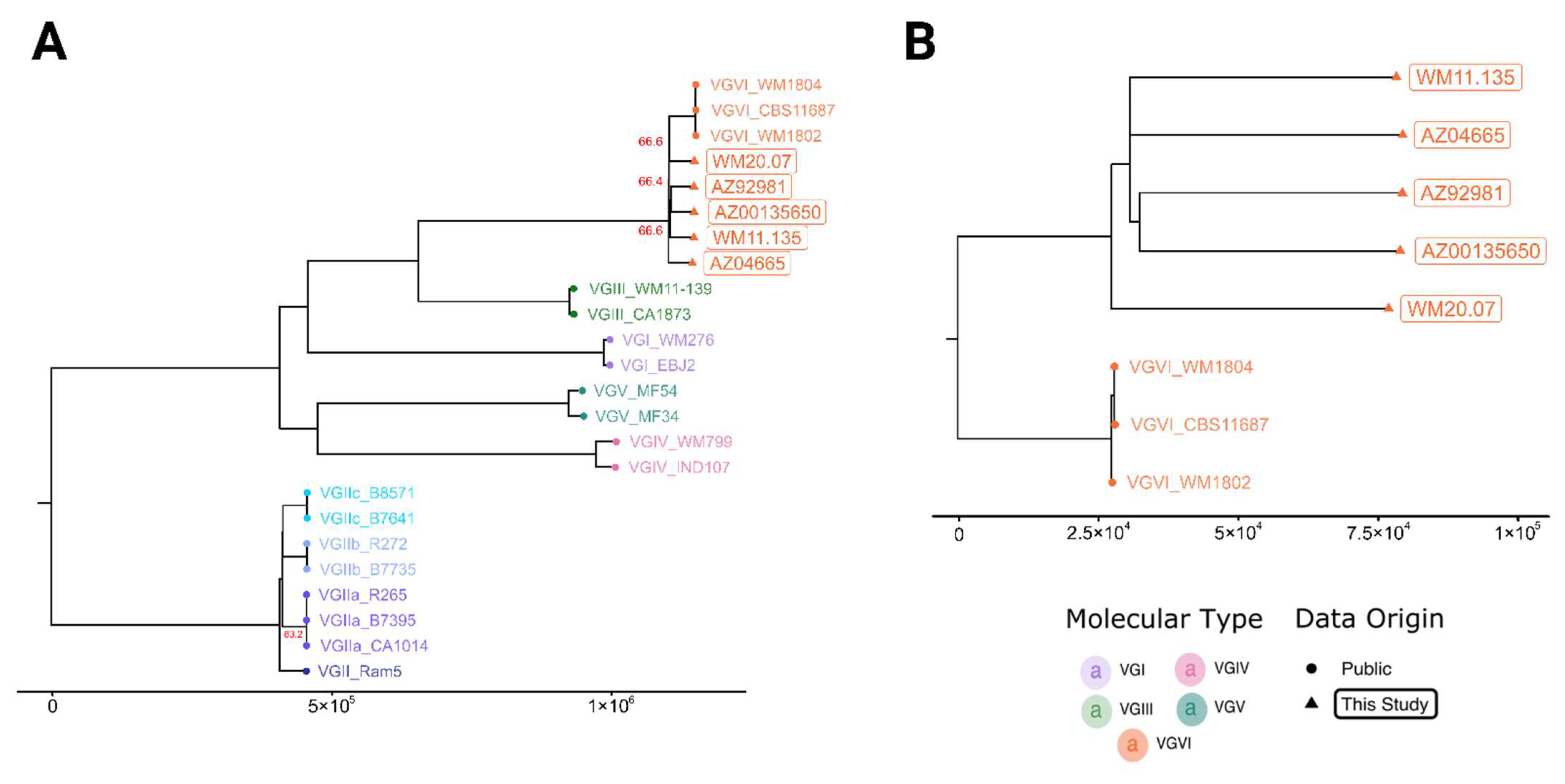{kind=link}
| ID | Isolate Source | Collection Locale | Collection Year | Sample Type | Comorbidity | Cryptococcosis Presentation | WGS Source | Metadata Source |
|---|---|---|---|---|---|---|---|---|
| AZ04665 | Clinical | USA/AZ | 2019 | CSF | HIV/AIDS | Meningitis | This Study | This study |
| AZ92981 | Clinical | USA/AZ | 2019 | Blood | Alcoholic cirrhosis and hepatitis, CHF | Pulmonary | This study | This study |
| AZ00135650 | Clinical | USA/AZ | 2021 | CSF | Pulmonary lesions/ Hx of Lymphoma | Bronchitis/ Meningitis | This study | This study |
| WM 11.135 | Veterinary | USA/AZ | 2011 | Nasal | Possible underlying hepatopathy | Upper respiratory signs | This study | This study |
| WM 20.07 | Clinical | Argentina/ Salta | 2017 | CSF | Malnutrition | Meningo- encephalitis | This study | [8] |
| WM 18.02 | Clinical | Mexico/DF | 1961 | CSF | ND | Meningitis | [4] | [7] |
| WM 18.04 | Clinical | Mexico/DF | 1965 | CSF | ND | Meningitis | [4] | [7] |
| CBS 11687 | Clinical | Mexico * | 1987 | ND | ND | ND | [6] | [9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monroy-Nieto, J.; Bowers, J.R.; Montfort, P.; Adame, G.; Taverna, C.G.; Yaglom, H.; Sykes, J.E.; Brady, S.; Mochon, A.B.; Meyer, W.; et al. Phylogenomic Placement of American Southwest-Associated Clinical and Veterinary Isolates Expands Evidence for Distinct Cryptococcus gattii VGVI. Microorganisms 2022, 10, 1681. https://doi.org/10.3390/microorganisms10081681
Monroy-Nieto J, Bowers JR, Montfort P, Adame G, Taverna CG, Yaglom H, Sykes JE, Brady S, Mochon AB, Meyer W, et al. Phylogenomic Placement of American Southwest-Associated Clinical and Veterinary Isolates Expands Evidence for Distinct Cryptococcus gattii VGVI. Microorganisms. 2022; 10(8):1681. https://doi.org/10.3390/microorganisms10081681
Chicago/Turabian StyleMonroy-Nieto, Juan, Jolene R. Bowers, Parker Montfort, Guillermo Adame, Constanza Giselle Taverna, Hayley Yaglom, Jane E. Sykes, Shane Brady, A. Brian Mochon, Wieland Meyer, and et al. 2022. "Phylogenomic Placement of American Southwest-Associated Clinical and Veterinary Isolates Expands Evidence for Distinct Cryptococcus gattii VGVI" Microorganisms 10, no. 8: 1681. https://doi.org/10.3390/microorganisms10081681
APA StyleMonroy-Nieto, J., Bowers, J. R., Montfort, P., Adame, G., Taverna, C. G., Yaglom, H., Sykes, J. E., Brady, S., Mochon, A. B., Meyer, W., Komatsu, K., & Engelthaler, D. M. (2022). Phylogenomic Placement of American Southwest-Associated Clinical and Veterinary Isolates Expands Evidence for Distinct Cryptococcus gattii VGVI. Microorganisms, 10(8), 1681. https://doi.org/10.3390/microorganisms10081681