
Human Cytomegalovirus and Human Herpesvirus 6 Coinfection of Dermal Fibroblasts Enhances the Pro-Inflammatory Pathway Predisposing to Fibrosis: The Possible Impact on Systemic Sclerosis

 , ,
, ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Cultures and Virus Strains
2.2. Viral Infection
2.3. Nucleic Acid Extraction
2.4. DNA Analysis: Virus Quantitation in Infected Cells
2.5. RNA Analysis
2.6. Statistical Analyses
3. Results
3.1. HCMV and HHV-6A Coinfection of Primary Human Dermal Fibroblasts
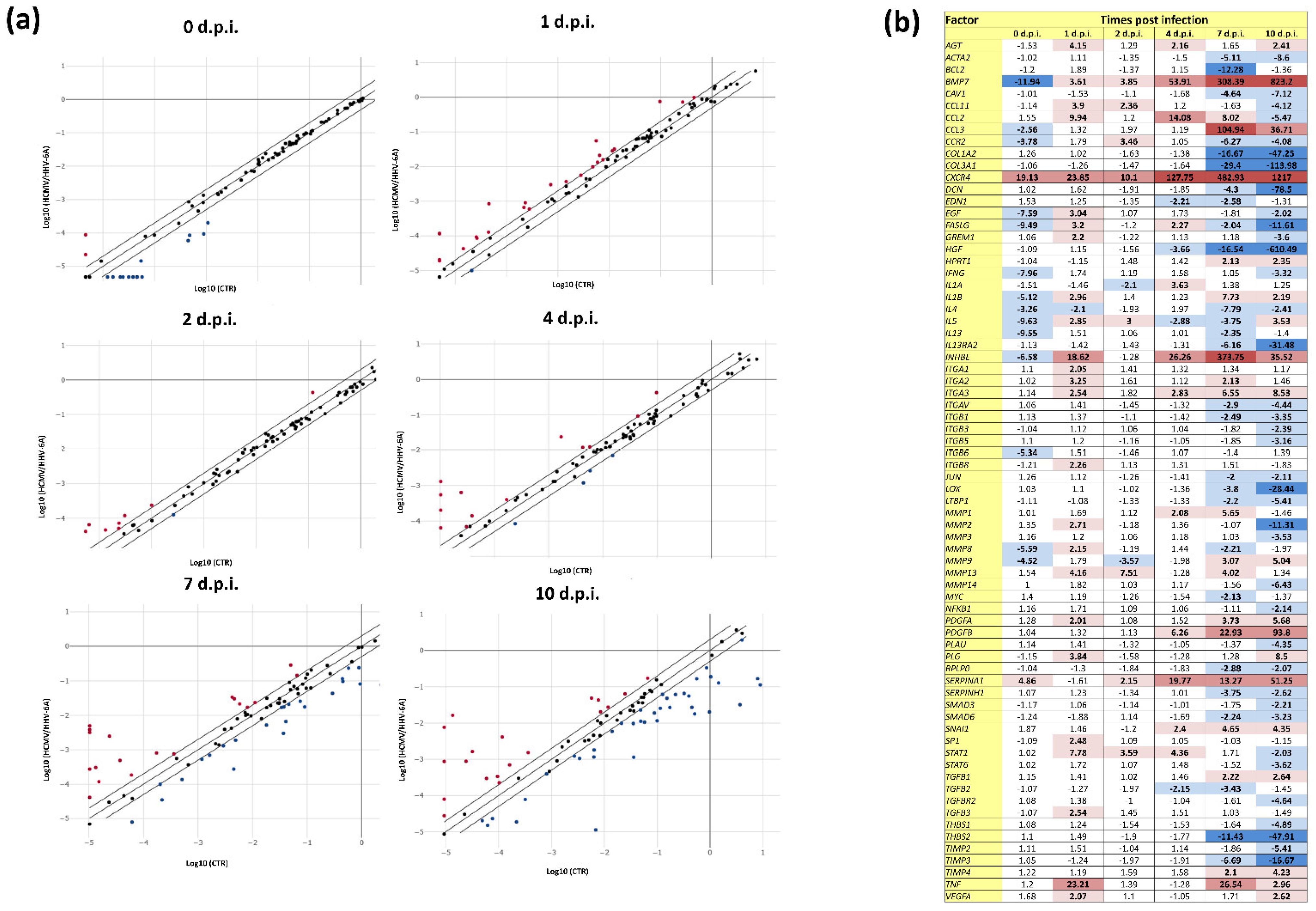
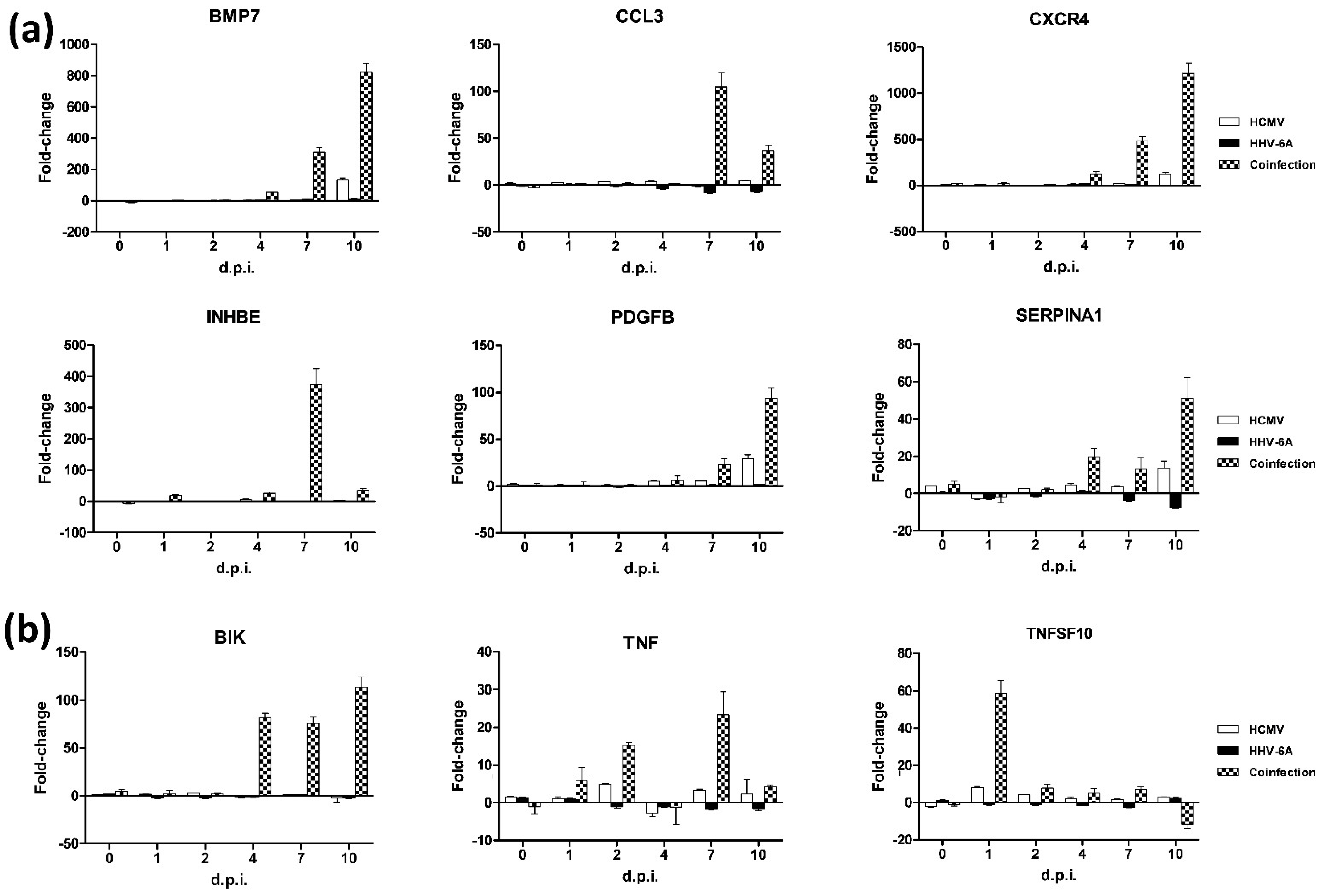
3.2. Effect of HCMV and HHV-6A Coinfection on the Expression of Fibrosis-Associated Factors
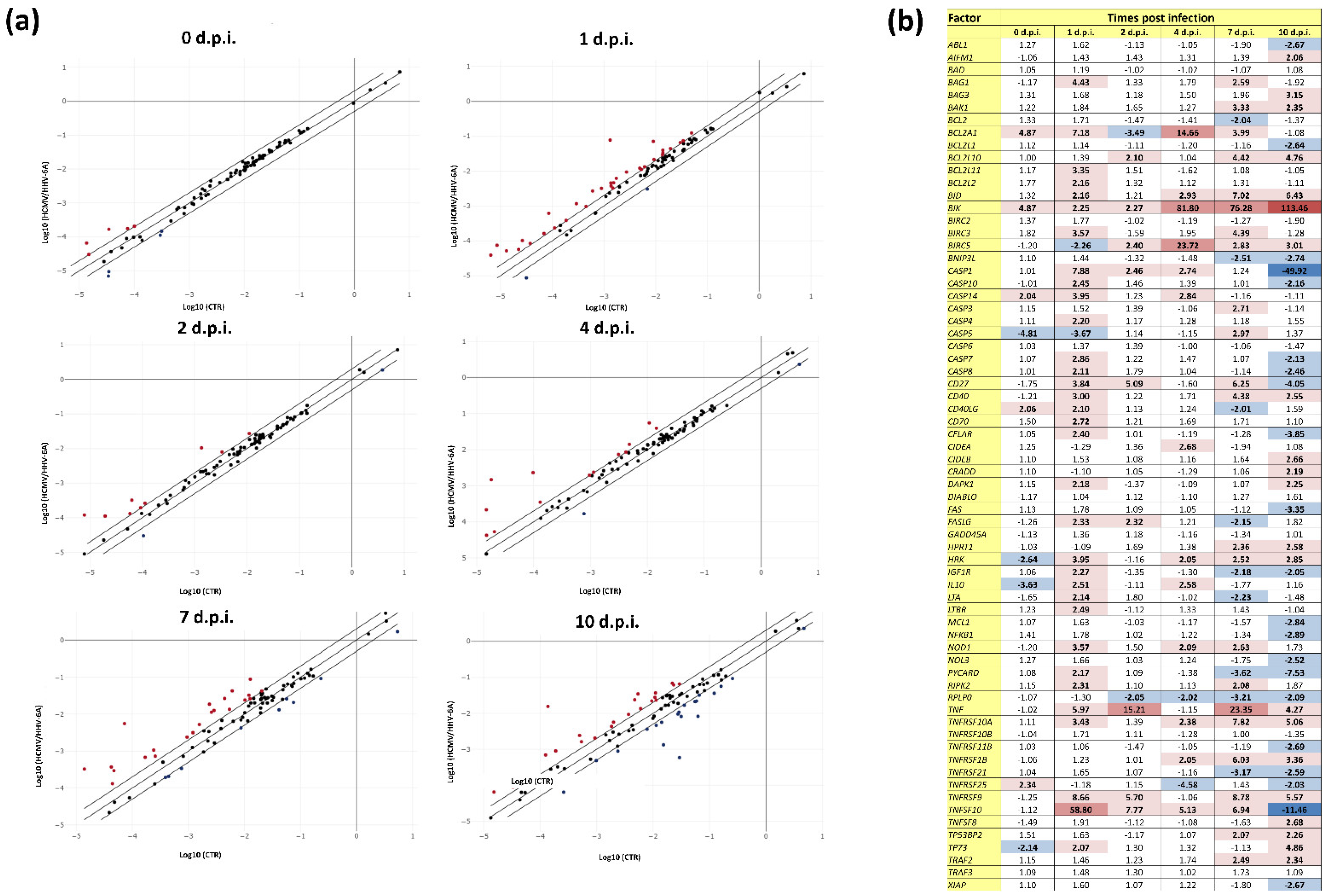
3.3. Effect of HCMV and HHV-6A Coinfection on the Expression of Apoptosis-Associated Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hinchcliff, M.; Mahoney, J.M. Towards a new classification of systemic sclerosis. Nat. Rev. Rheumatol. 2019, 15, 456–457. [Google Scholar] [CrossRef] [PubMed]
- Lescoat, A.; Roofeh, D.; Kuwana, M.; Lafyatis, R.; Allanore, Y.; Khanna, D. Therapeutic approaches to systemic sclerosis: Recent approvals and future candidate therapies. Clin. Rev. Allergy Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.; Arcangeletti, M.C.; Caselli, E.; Zakrzewska, K.; Maccari, C.; Calderaro, A.; D’Accolti, M.; Soffritti, I.; Arvia, R.; Sighinolfi, G.; et al. Insights into the knowledge of complex diseases: Environmental infectious/toxic agents as potential etiopathogenetic factors of systemic sclerosis. J. Autoimmun. 2021, 124, 102727. [Google Scholar] [CrossRef] [PubMed]
- Chabaud, S.; Moulin, V.J. Apoptosis modulation as a promising target for treatment of systemic sclerosis. Int. J. Rheumatol. 2011, 2011, 495792. [Google Scholar] [CrossRef]
- Ates, A.; Kinikli, G.; Turgay, M.; Duman, M. The levels of serum-soluble fas in patients with rheumatoid arthritis and systemic sclerosis. Clin. Rheumatol. 2004, 23, 421–425. [Google Scholar] [CrossRef]
- Santiago, B.; Galindo, M.; Rivero, M.; Pablos, J.L. Decreased susceptibility to fas-induced apoptosis of systemic sclerosis dermal fibroblasts. Arthritis Rheum. 2001, 44, 1667–1676. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yokozeki, H.; Nishioka, K. Fas- and fasl-deficient mice are resistant to the induction of bleomycin-induced scleroderma. Arch. Dermatol. Res. 2007, 298, 465–468. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nishioka, K. Possible role of apoptosis in the pathogenesis of bleomycin-induced scleroderma. J. Investig. Dermatol. 2004, 122, 44–50. [Google Scholar] [CrossRef]
- Lunardi, C.; Bason, C.; Navone, R.; Millo, E.; Damonte, G.; Corrocher, R.; Puccetti, A. Systemic sclerosis immunoglobulin g autoantibodies bind the human cytomegalovirus late protein ul94 and induce apoptosis in human endothelial cells. Nat. Med. 2000, 6, 1183–1186. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Tan, F.K.; Arnett, F.C.; Jin, L.; Geng, Y.J. Induction of apoptosis and fibrillin 1 expression in human dermal endothelial cells by scleroderma sera containing anti-endothelial cell antibodies. Arthritis Rheum. 2006, 54, 2250–2262. [Google Scholar] [CrossRef]
- Zhang, X.L.; Topley, N.; Ito, T.; Phillips, A. Interleukin-6 regulation of transforming growth factor (tgf)-beta receptor compartmentalization and turnover enhances tgf-beta1 signaling. J. Biol. Chem. 2005, 280, 12239–12245. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y.P.; Misso, N.L.; Scaffidi, A.K.; Fogel-Petrovic, M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am. J. Respir. Cell Mol. Biol. 2003, 29, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Arcangeletti, M.C.; D’Accolti, M.; Maccari, C.; Soffritti, I.; Conto, F.; Chezzi, C.; Calderaro, A.; Ferri, C.; Caselli, E. Impact of human cytomegalovirus and human herpesvirus 6 infection on the expression of factors associated with cell fibrosis and apoptosis: Clues for implication in systemic sclerosis development. Int. J. Mol. Sci. 2020, 21, 6397. [Google Scholar] [CrossRef] [PubMed]
- Arcangeletti, M.C.; Maccari, C.; Vescovini, R.; Volpi, R.; Giuggioli, D.; Sighinolfi, G.; De Conto, F.; Chezzi, C.; Calderaro, A.; Ferri, C. A paradigmatic interplay between human cytomegalovirus and host immune system: Possible involvement of viral antigen-driven cd8+ t cell responses in systemic sclerosis. Viruses 2018, 10, 508. [Google Scholar] [CrossRef]
- Caselli, E.; Soffritti, I.; D’Accolti, M.; Bortolotti, D.; Rizzo, R.; Sighinolfi, G.; Giuggioli, D.; Ferri, C. Hhv-6a infection and systemic sclerosis: Clues of a possible association. Microorganisms 2019, 8, 39. [Google Scholar] [CrossRef]
- Broccolo, F.; Drago, F.; Cassina, G.; Fava, A.; Fusetti, L.; Matteoli, B.; Ceccherini-Nelli, L.; Sabbadini, M.G.; Lusso, P.; Parodi, A.; et al. Selective reactivation of human herpesvirus 6 in patients with autoimmune connective tissue diseases. J. Med. Virol. 2013, 85, 1925–1934. [Google Scholar] [CrossRef]
- Broccolo, F.; Drago, F.; Paolino, S.; Cassina, G.; Gatto, F.; Fusetti, L.; Matteoli, B.; Zaccaria, E.; Parodi, A.; Lusso, P.; et al. Reactivation of human herpesvirus 6 (hhv-6) infection in patients with connective tissue diseases. J. Clin. Virol. 2009, 46, 43–46. [Google Scholar] [CrossRef]
- Zerr, D.M.; Meier, A.S.; Selke, S.S.; Frenkel, L.M.; Huang, M.L.; Wald, A.; Rhoads, M.P.; Nguy, L.; Bornemann, R.; Morrow, R.A.; et al. A population-based study of primary human herpesvirus 6 infection. N. Engl. J. Med. 2005, 352, 768–776. [Google Scholar] [CrossRef]
- Pass, R.F. Epidemiology and transmission of cytomegalovirus. J. Infect. Dis. 1985, 152, 243–248. [Google Scholar] [CrossRef]
- Sehrawat, S.; Kumar, D.; Rouse, B.T. Herpesviruses: Harmonious pathogens but relevant cofactors in other diseases? Front. Cell Infect. Microbiol. 2018, 8, 177. [Google Scholar] [CrossRef]
- Caselli, E.; Di Luca, D. Molecular biology and clinical associations of roseoloviruses human herpesvirus 6 and human herpesvirus 7. New Microbiol. 2007, 30, 173–187. [Google Scholar] [PubMed]
- Broccolo, F.; Fusetti, L.; Ceccherini-Nelli, L. Possible role of human herpesvirus 6 as a trigger of autoimmune disease. Sci. World J. 2013, 2013, 867389. [Google Scholar] [CrossRef]
- Ranger-Rogez, S.; Vidal, E.; Liozon, F.; Denis, F. Primary sjogren’s syndrome and antibodies to human herpesvirus type 6. Clin. Infect. Dis. 1994, 19, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Krueger, G.R.; Sander, C.; Hoffmann, A.; Barth, A.; Koch, B.; Braun, M. Isolation of human herpesvirus-6 (hhv-6) from patients with collagen vascular diseases. In Vivo 1991, 5, 217–225. [Google Scholar] [PubMed]
- Alvarez-Lafuente, R.; Fernandez-Gutierrez, B.; de Miguel, S.; Jover, J.A.; Rollin, R.; Loza, E.; Clemente, D.; Lamas, J.R. Potential relationship between herpes viruses and rheumatoid arthritis: Analysis with quantitative real time polymerase chain reaction. Ann. Rheum. Dis. 2005, 64, 1357–1359. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.; Cazzato, M.; Giuggioli, D.; Sebastiani, M.; Magro, C. Systemic sclerosis following human cytomegalovirus infection. Ann. Rheum. Dis. 2002, 61, 937–938. [Google Scholar] [CrossRef]
- Lunardi, C.; Dolcino, M.; Peterlana, D.; Bason, C.; Navone, R.; Tamassia, N.; Beri, R.; Corrocher, R.; Puccetti, A. Antibodies against human cytomegalovirus in the pathogenesis of systemic sclerosis: A gene array approach. PLoS Med. 2006, 3, e2. [Google Scholar] [CrossRef]
- Arnson, Y.; Amital, H.; Guiducci, S.; Matucci-Cerinic, M.; Valentini, G.; Barzilai, O.; Maya, R.; Shoenfeld, Y. The role of infections in the immunopathogensis of systemic sclerosis--evidence from serological studies. Ann. N. Y. Acad. Sci. 2009, 1173, 627–632. [Google Scholar] [CrossRef]
- Marou, E.; Liaskos, C.; Simopoulou, T.; Efthymiou, G.; Dardiotis, E.; Katsiari, C.; Scheper, T.; Meyer, W.; Hadjigeorgiou, G.; Bogdanos, D.P.; et al. Human cytomegalovirus (hcmv) ul44 and ul57 specific antibody responses in anti-hcmv-positive patients with systemic sclerosis. Clin. Rheumatol. 2017, 36, 863–869. [Google Scholar] [CrossRef]
- Efthymiou, G.; Dardiotis, E.; Liaskos, C.; Marou, E.; Scheper, T.; Meyer, W.; Daponte, A.; Daoussis, D.; Hadjigeorgiou, G.; Bogdanos, D.P.; et al. A comprehensive analysis of antigen-specific antibody responses against human cytomegalovirus in patients with systemic sclerosis. Clin. Immunol. 2019, 207, 87–96. [Google Scholar] [CrossRef]
- Caselli, E.; Zatelli, M.C.; Rizzo, R.; Benedetti, S.; Martorelli, D.; Trasforini, G.; Cassai, E.; degli Uberti, E.C.; Di Luca, D.; Dolcetti, R. Virologic and immunologic evidence supporting an association between hhv-6 and hashimoto’s thyroiditis. PLoS Pathog. 2012, 8, e1002951. [Google Scholar] [CrossRef] [PubMed]
- Halenius, A.; Hengel, H. Human cytomegalovirus and autoimmune disease. BioMed. Res. Int. 2014, 2014, 472978. [Google Scholar] [CrossRef]
- Barsotti, S.; Orlandi, M.; Codullo, V.; Di Battista, M.; Lepri, G.; Della Rossa, A.; Guiducci, S. One year in review 2019: Systemic sclerosis. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S119), 3–14. [Google Scholar] [PubMed]
- Sinzger, C.; Grefte, A.; Plachter, B.; Gouw, A.S.; The, T.H.; Jahn, G. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 1995, 76 Pt 4, 741–750. [Google Scholar] [CrossRef]
- Mostmans, Y.; Cutolo, M.; Giddelo, C.; Decuman, S.; Melsens, K.; Declercq, H.; Vandecasteele, E.; De Keyser, F.; Distler, O.; Gutermuth, J.; et al. The role of endothelial cells in the vasculopathy of systemic sclerosis: A systematic review. Autoimmun. Rev. 2017, 16, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Soffritti, I.; D’Accolti, M.; Ravegnini, G.; Arcangeletti, M.C.; Maccari, C.; De Conto, F.; Calderaro, A.; Caselli, E. Modulation of micrornome by human cytomegalovirus and human herpesvirus 6 infection in human dermal fibroblasts: Possible significance in the induction of fibrosis in systemic sclerosis. Cells 2021, 10, 1060. [Google Scholar] [CrossRef]
- DesJardin, J.A.; Gibbons, L.; Cho, E.; Supran, S.E.; Falagas, M.E.; Werner, B.G.; Snydman, D.R. Human herpesvirus 6 reactivation is associated with cytomegalovirus infection and syndromes in kidney transplant recipients at risk for primary cytomegalovirus infection. J. Infect. Dis. 1998, 178, 1783–1786. [Google Scholar] [CrossRef][Green Version]
- Van Leer-Buter, C.C.; Sanders, J.S.; Vroom, H.E.; Riezebos-Brilman, A.; Niesters, H.G. Human herpesvirus-6 dnaemia is a sign of impending primary cmv infection in cmv sero-discordant renal transplantations. J. Clin. Virol. 2013, 58, 422–426. [Google Scholar] [CrossRef]
- Handous, I.; Achour, B.; Marzouk, M.; Rouis, S.; Hazgui, O.; Brini, I.; Khelif, A.; Hannachi, N.; Boukadida, J. Co-infections of human herpesviruses (cmv, hhv-6, hhv-7 and ebv) in non-transplant acute leukemia patients undergoing chemotherapy. Virol. J. 2020, 17, 37. [Google Scholar] [CrossRef]
- Lopez Roa, P.; Hill, J.A.; Kirby, K.A.; Leisenring, W.M.; Huang, M.L.; Santo, T.K.; Jerome, K.R.; Boeckh, M.; Limaye, A.P. Coreactivation of human herpesvirus 6 and cytomegalovirus is associated with worse clinical outcome in critically ill adults. Crit. Care Med. 2015, 43, 1415–1422. [Google Scholar] [CrossRef]
- Doridot, L.; Jeljeli, M.; Chene, C.; Batteux, F. Implication of oxidative stress in the pathogenesis of systemic sclerosis via inflammation, autoimmunity and fibrosis. Redox Biol. 2019, 25, 101122. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Bracci, A.; Galvan, M.; Boni, M.; Rotola, A.; Bergamini, C.; Cermelli, C.; Dal Monte, P.; Gompels, U.A.; Cassai, E.; et al. Human herpesvirus 6 (hhv-6) u94/rep protein inhibits betaherpesvirus replication. Virology 2006, 346, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, P.; Franca Milia, A.; Liakouli, V.; Pacini, A.; Manetti, M.; Marrelli, A.; Toscano, A.; Pingiotti, E.; Fulminis, A.; Guiducci, S.; et al. Differential expression of stromal cell-derived factor 1 and its receptor cxcr4 in the skin and endothelial cells of systemic sclerosis patients: Pathogenetic implications. Arthritis Rheum. 2006, 54, 3022–3033. [Google Scholar] [CrossRef]
- Li, F.; Xu, X.; Geng, J.; Wan, X.; Dai, H. The autocrine cxcr4/cxcl12 axis contributes to lung fibrosis through modulation of lung fibroblast activity. Exp. Ther. Med. 2020, 19, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Brietzke, A.; Guthoff, R.; Stahnke, T.; Grabow, N. Fibrosis: Altered gene expression in tgf-β stimulated human fibroblasts of the tenon. Curr. Dir. Biomed. Eng. 2020, 6, 430–433. [Google Scholar] [CrossRef]
- Sun, Y.; Cai, H.; Ge, J.; Shao, F.; Huang, Z.; Ding, Z.; Dong, L.; Chen, J.; Zhang, J.; Zang, Y. Tubule-derived inhbb promotes interstitial fibroblast activation and renal fibrosis. J. Pathol. 2022, 256, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, L.M.; Hill, C.S. Beyond tgfbeta: Roles of other tgfbeta superfamily members in cancer. Nat. Rev. Cancer 2013, 13, 328–341. [Google Scholar] [CrossRef]
- Makanji, Y.; Zhu, J.; Mishra, R.; Holmquist, C.; Wong, W.P.; Schwartz, N.B.; Mayo, K.E.; Woodruff, T.K. Inhibin at 90: From discovery to clinical application, a historical review. Endocr. Rev. 2014, 35, 747–794. [Google Scholar] [CrossRef]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2010, 147, 35–51. [Google Scholar] [CrossRef]
- Corradini, E.; Babitt, J.L.; Lin, H.Y. The rgm/dragon family of bmp co-receptors. Cytokine Growth Factor. Rev. 2009, 20, 389–398. [Google Scholar] [CrossRef]
- Li, R.X.; Yiu, W.H.; Tang, S.C. Role of bone morphogenetic protein-7 in renal fibrosis. Front. Physiol. 2015, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; An, G.; Wang, Y.; Liang, D.; Zhu, Z.; Lian, X.; Niu, P.; Guo, C.; Tian, L. Anti-fibrotic effects of bone morphogenetic protein-7-modified bone marrow mesenchymal stem cells on silica-induced pulmonary fibrosis. Exp. Mol. Pathol. 2017, 102, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Bandinelli, F.; Del Rosso, A.; Gabrielli, A.; Giacomelli, R.; Bartoli, F.; Guiducci, S.; Matucci Cerinic, M. Ccl2, ccl3 and ccl5 chemokines in systemic sclerosis: The correlation with ssc clinical features and the effect of prostaglandin e1 treatment. Clin. Exp. Rheumatol. 2012, 30, S44–S49. [Google Scholar]
- Goncalves, R.S.G.; Pereira, M.C.; Dantas, A.T.; Almeida, A.R.; Rego, M.; Lima, E.A.; Pitta, I.D.R.; Duarte, A.; Pitta, M. Ccl3, il-7, il-13 and ifngamma transcripts are increased in skin’s biopsy of systemic sclerosis. Exp. Dermatol. 2019, 28, 1172–1175. [Google Scholar] [CrossRef]
- Huaux, F.; Gharaee-Kermani, M.; Liu, T.; Morel, V.; McGarry, B.; Ullenbruch, M.; Kunkel, S.L.; Wang, J.; Xing, Z.; Phan, S.H. Role of eotaxin-1 (ccl11) and cc chemokine receptor 3 (ccr3) in bleomycin-induced lung injury and fibrosis. Am. J. Pathol. 2005, 167, 1485–1496. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Pilling, D.; Vakil, V.; Cox, N.; Gomer, R.H. Tnf-alpha-stimulated fibroblasts secrete lumican to promote fibrocyte differentiation. Proc Natl Acad Sci. USA 2015, 112, 11929–11934. [Google Scholar] [CrossRef]
- Taguchi, S.; Azushima, K.; Yamaji, T.; Urate, S.; Suzuki, T.; Abe, E.; Tanaka, S.; Tsukamoto, S.; Kamimura, D.; Kinguchi, S.; et al. Effects of tumor necrosis factor-alpha inhibition on kidney fibrosis and inflammation in a mouse model of aristolochic acid nephropathy. Sci. Rep. 2021, 11, 23587. [Google Scholar] [CrossRef]
- Lam, G.K.; Hummers, L.K.; Woods, A.; Wigley, F.M. Efficacy and safety of etanercept in the treatment of scleroderma-associated joint disease. J. Rheumatol. 2007, 34, 1636–1637. [Google Scholar]
- Bosello, S.; De Santis, M.; Tolusso, B.; Zoli, A.; Ferraccioli, G. Tumor necrosis factor-alpha inhibitor therapy in erosive polyarthritis secondary to systemic sclerosis. Ann. Intern. Med. 2005, 143, 918–920. [Google Scholar] [CrossRef]
- Clements, P.; Lachenbruch, P.; Siebold, J.; White, B.; Weiner, S.; Martin, R.; Weinstein, A.; Weisman, M.; Mayes, M.; Collier, D.; et al. Inter and intraobserver variability of total skin thickness score (modified rodnan tss) in systemic sclerosis. J. Rheumatol. 1995, 22, 1281–1285. [Google Scholar] [PubMed]
- Distler, J.H.; Schett, G.; Gay, S.; Distler, O. The controversial role of tumor necrosis factor alpha in fibrotic diseases. Arthritis Rheum. 2008, 58, 2228–2235. [Google Scholar] [CrossRef]
- Trojanowska, M. Role of pdgf in fibrotic diseases and systemic sclerosis. Rheumatology 2008, 47 (Suppl. S5), v2–v4. [Google Scholar] [CrossRef]
- Zhang, W.; Ge, Y.; Cheng, Q.; Zhang, Q.; Fang, L.; Zheng, J. Decorin is a pivotal effector in the extracellular matrix and tumour microenvironment. Oncotarget 2018, 9, 5480–5491. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, R.V.; Worrell, J.C.; Boylan, D.; Walsh, S.M.; Cramton, J.; Counihan, I.; O’Beirne, S.; Medina, M.F.; Gauldie, J.; Fabre, A.; et al. Modulation of pulmonary fibrosis by il-13ralpha2. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L710–L718. [Google Scholar] [CrossRef] [PubMed]
- Leong, E.; Bezuhly, M.; Marshall, J.S. Distinct metalloproteinase expression and functions in systemic sclerosis and fibrosis: What we know and the potential for intervention. Front. Physiol. 2021, 12, 727451. [Google Scholar] [CrossRef]
- Niwa, H.; Kanno, Y.; Shu, E.; Seishima, M. Decrease in matrix metalloproteinase3 activity in systemic sclerosis fibroblasts causes alpha2antiplasmin and extracellular matrix deposition, and contributes to fibrosis development. Mol. Med. Rep. 2020, 22, 3001–3007. [Google Scholar] [PubMed]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Model Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef]
- Russo, B.; Brembilla, N.C.; Chizzolini, C. Interplay between keratinocytes and fibroblasts: A systematic review providing a new angle for understanding skin fibrotic disorders. Front Immunol. 2020, 11, 648. [Google Scholar] [CrossRef]
- Feng, D.; Gerarduzzi, C. Emerging roles of matricellular proteins in systemic sclerosis. Int. J. Mol. Sci. 2020, 21, 4776. [Google Scholar] [CrossRef]
- Kopinski, P.; Chorostowska, J.; Przybylski, G.; Dyczek, A.; Wedrowska, E.; Jankowski, M.; Szpechcinski, A.; Gizycka, A.; Golinska, J. Studies on hepatocyte growth factor (hgf) in bronchoalveolar lavage (bal) do not support the concept on its antifibrotic properties in idiopathic pulmonary fibrosis (ipf). Eur. Respir. J. 2015, 46, PA3850. [Google Scholar]
- Frost, J.; Ramsay, M.; Mia, R.; Moosa, L.; Musenge, E.; Tikly, M. Differential gene expression of mmp-1, timp-1 and hgf in clinically involved and uninvolved skin in south africans with ssc. Rheumatology 2012, 51, 1049–1052. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Harigai, M.; Hara, M.; Fukasawa, C.; Takagi, K.; Tanaka, M.; Tanaka, E.; Nishimagi, E.; Kamatani, N. Expression of hepatocyte growth factor and its receptor (c-met) in skin fibroblasts from patients with systemic sclerosis. J. Rheumatol. 2002, 29, 1877–1883. [Google Scholar] [PubMed]
- Dosreis, G.A.; Borges, V.M.; Zin, W.A. The central role of fas-ligand cell signaling in inflammatory lung diseases. J. Cell Mol. Med. 2004, 8, 285–293. [Google Scholar] [CrossRef]
- Sawamura, S.; Makino, K.; Ide, M.; Shimada, S.; Kajihara, I.; Makino, T.; Jinnin, M.; Fukushima, S. Elevated alpha 1(i) to alpha 2(i) collagen ratio in dermal fibroblasts possibly contributes to fibrosis in systemic sclerosis. Int. J. Mol. Sci. 2022, 23, 6811. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; Hock, T.; Thannickal, V.J.; Sanders, Y.Y. Histone deacetylase inhibition downregulates collagen 3a1 in fibrotic lung fibroblasts. Int. J. Mol. Sci. 2013, 14, 19605–19617. [Google Scholar] [CrossRef]
- Nguyen, X.X.; Nishimoto, T.; Takihara, T.; Mlakar, L.; Bradshaw, A.D.; Feghali-Bostwick, C. Lysyl oxidase directly contributes to extracellular matrix production and fibrosis in systemic sclerosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 320, L29–L40. [Google Scholar] [CrossRef]
- Xu, J.D.; Cao, X.X.; Long, Z.W.; Liu, X.P.; Furuya, T.; Xu, J.W.; Liu, X.L.; De Xu, Z.; Sasaki, K.; Li, Q.Q. Bcl2l10 protein regulates apoptosis/proliferation through differential pathways in gastric cancer cells. J. Pathol. 2011, 223, 400–409. [Google Scholar] [CrossRef]
- Renshaw, S.A.; Dempsey, C.E.; Barnes, F.A.; Bagstaff, S.M.; Dower, S.K.; Bingle, C.D.; Whyte, M.K. Three novel bid proteins generated by alternative splicing of the human bid gene. J. Biol. Chem. 2004, 279, 2846–2855. [Google Scholar] [CrossRef]
- Chinnadurai, G.; Vijayalingam, S.; Rashmi, R. Bik, the founding member of the bh3-only family proteins: Mechanisms of cell death and role in cancer and pathogenic processes. Oncogene 2008, 27 (Suppl. S1), S20–S29. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Davies, C.; Knox, P.G.; Gallagher, N.J.; Afford, S.C.; Adams, D.H.; Young, L.S. Cd40 induces apoptosis in carcinoma cells through activation of cytotoxic ligands of the tumor necrosis factor superfamily. Mol. Cell Biol. 2000, 20, 5503–5515. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-based propagation of functional annotations within the gene ontology consortium. Brief Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Rath, P.C.; Aggarwal, B.B. Tnf-induced signaling in apoptosis. J. Clin. Immunol. 1999, 19, 350–364. [Google Scholar] [CrossRef]
- Li, T.; Su, L.; Lei, Y.; Liu, X.; Zhang, Y.; Liu, X. Ddit3 and kat2a proteins regulate tnfrsf10a and tnfrsf10b expression in endoplasmic reticulum stress-mediated apoptosis in human lung cancer cells. J. Biol. Chem. 2015, 290, 11108–11118. [Google Scholar] [CrossRef]
- Messeha, S.S.; Zarmouh, N.O.; Mendonca, P.; Alwagdani, H.; Cotton, C.; Soliman, K.F.A. Effects of gossypol on apoptosisrelated gene expression in racially distinct triplenegative breast cancer cells. Oncol. Rep. 2019, 42, 467–478. [Google Scholar]
- Frazzi, R. Birc3 and birc5: Multi-faceted inhibitors in cancer. Cell Biosci. 2021, 11, 8. [Google Scholar] [CrossRef]
- Jun, J.B.; Kuechle, M.; Harlan, J.M.; Elkon, K.B. Fibroblast and endothelial apoptosis in systemic sclerosis. Curr. Opin. Rheumatol. 2003, 15, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.B.; Farashahi Yazd, E.; Gharibdoost, F.; Sheikhha, M.H.; Karimizadeh, E.; Jamshidi, A.; Mahmoudi, M. Overexpression of apoptosis-related protein, survivin, in fibroblasts from patients with systemic sclerosis. Ir. J. Med. Sci. 2019, 188, 1443–1449. [Google Scholar] [CrossRef]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef]
- Herrera, B.; Alvarez, A.M.; Beltran, J.; Valdes, F.; Fabregat, I.; Fernandez, M. Resistance to tgf-beta-induced apoptosis in regenerating hepatocytes. J. Cell Physiol. 2004, 201, 385–392. [Google Scholar] [CrossRef]
- Salvesen, G.S.; Duckett, C.S. Iap proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Sisson, T.H.; Maher, T.M.; Ajayi, I.O.; King, J.E.; Higgins, P.D.; Booth, A.J.; Sagana, R.L.; Huang, S.K.; White, E.S.; Moore, B.B.; et al. Increased survivin expression contributes to apoptosis-resistance in ipf fibroblasts. Adv. Biosci. Biotechnol. 2012, 3, 657–664. [Google Scholar] [CrossRef] [PubMed][Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Single Infections | Coinfections | |||
|---|---|---|---|---|
| d.p.i. | HCMV (Copies/µg) 1 | HHV-6A (Copies/µg) 1 | HCMV (Copies/µg) 1 | HHV-6A (Copies/µg) 1 |
| 0 | - | - | - | - |
| 1 | 3.976 ± 0.002 | 5.012 ± 0.002 | 5.017 ± 0.005 | 5.560 ± 0.004 |
| 2 | 4.767 ± 0.003 | 5.340 ± 0.004 | 5.532 ± 0.004 | 6.227 ± 0.006 |
| 4 | 5.021 ± 0.001 | 5.745 ± 0.003 | 7.092 ± 0.003 | 7.307 ± 0.007 |
| 7 | 5.468 ± 0.005 | 4.993 ± 0.001 | 7.977 ± 0.002 | 5.748 ± 0.001 |
| 10 | 5.651 ± 0.004 | 4.750 ± 0.002 | 8.690 ± 0.004 | 5.470 ± 0.003 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soffritti, I.; D’Accolti, M.; Maccari, C.; Bini, F.; Mazziga, E.; de Conto, F.; Calderaro, A.; Arcangeletti, M.-C.; Caselli, E. Human Cytomegalovirus and Human Herpesvirus 6 Coinfection of Dermal Fibroblasts Enhances the Pro-Inflammatory Pathway Predisposing to Fibrosis: The Possible Impact on Systemic Sclerosis. Microorganisms 2022, 10, 1600. https://doi.org/10.3390/microorganisms10081600
Soffritti I, D’Accolti M, Maccari C, Bini F, Mazziga E, de Conto F, Calderaro A, Arcangeletti M-C, Caselli E. Human Cytomegalovirus and Human Herpesvirus 6 Coinfection of Dermal Fibroblasts Enhances the Pro-Inflammatory Pathway Predisposing to Fibrosis: The Possible Impact on Systemic Sclerosis. Microorganisms. 2022; 10(8):1600. https://doi.org/10.3390/microorganisms10081600
Chicago/Turabian StyleSoffritti, Irene, Maria D’Accolti, Clara Maccari, Francesca Bini, Eleonora Mazziga, Flora de Conto, Adriana Calderaro, Maria-Cristina Arcangeletti, and Elisabetta Caselli. 2022. "Human Cytomegalovirus and Human Herpesvirus 6 Coinfection of Dermal Fibroblasts Enhances the Pro-Inflammatory Pathway Predisposing to Fibrosis: The Possible Impact on Systemic Sclerosis" Microorganisms 10, no. 8: 1600. https://doi.org/10.3390/microorganisms10081600
APA StyleSoffritti, I., D’Accolti, M., Maccari, C., Bini, F., Mazziga, E., de Conto, F., Calderaro, A., Arcangeletti, M.-C., & Caselli, E. (2022). Human Cytomegalovirus and Human Herpesvirus 6 Coinfection of Dermal Fibroblasts Enhances the Pro-Inflammatory Pathway Predisposing to Fibrosis: The Possible Impact on Systemic Sclerosis. Microorganisms, 10(8), 1600. https://doi.org/10.3390/microorganisms10081600