
A Propidium Monoazide (PMAxx)-Droplet Digital PCR (ddPCR) for the Detection of Viable Burkholderia cepacia Complex in Nuclease-Free Water and Antiseptics

Abstract
:1. Introduction
2. Materials and Methods
2.1. ddPCR Assay
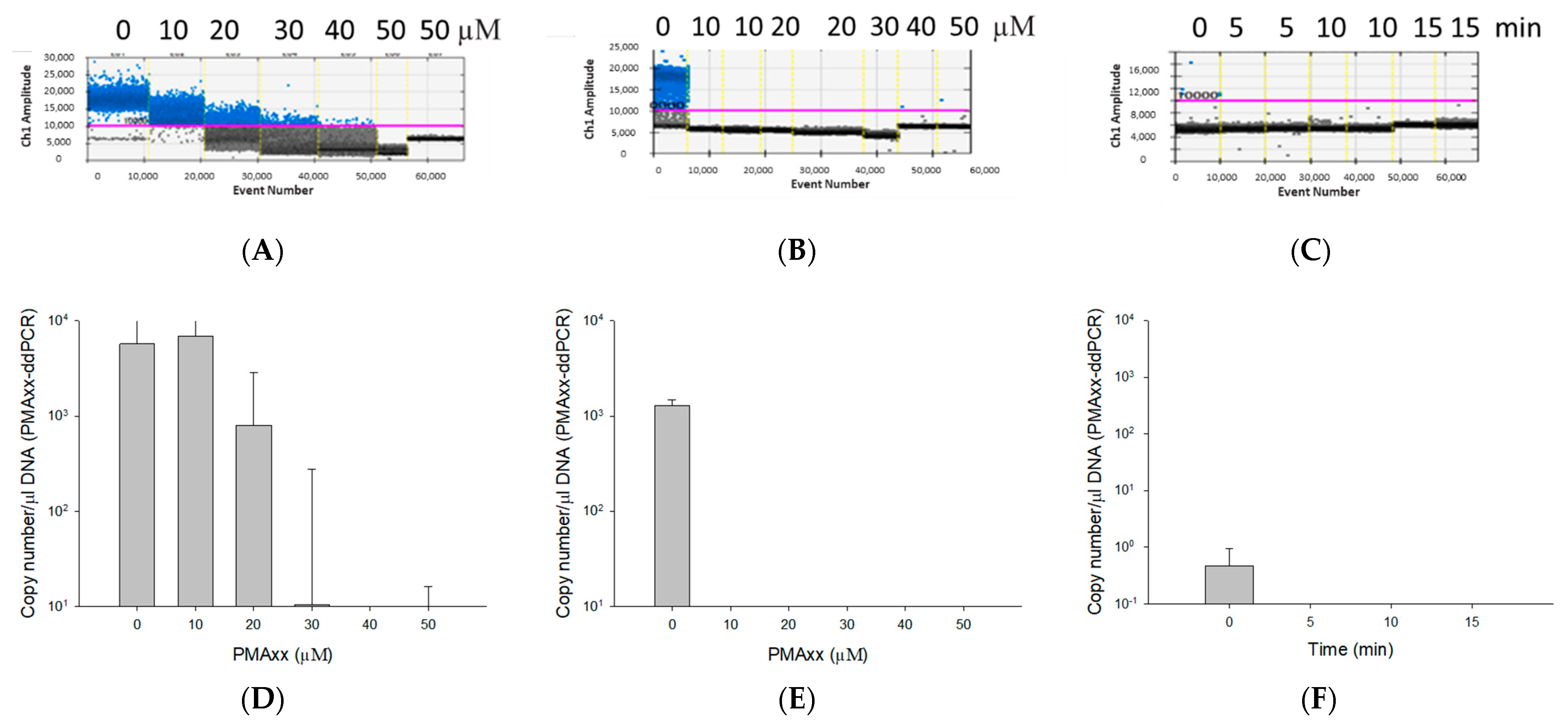
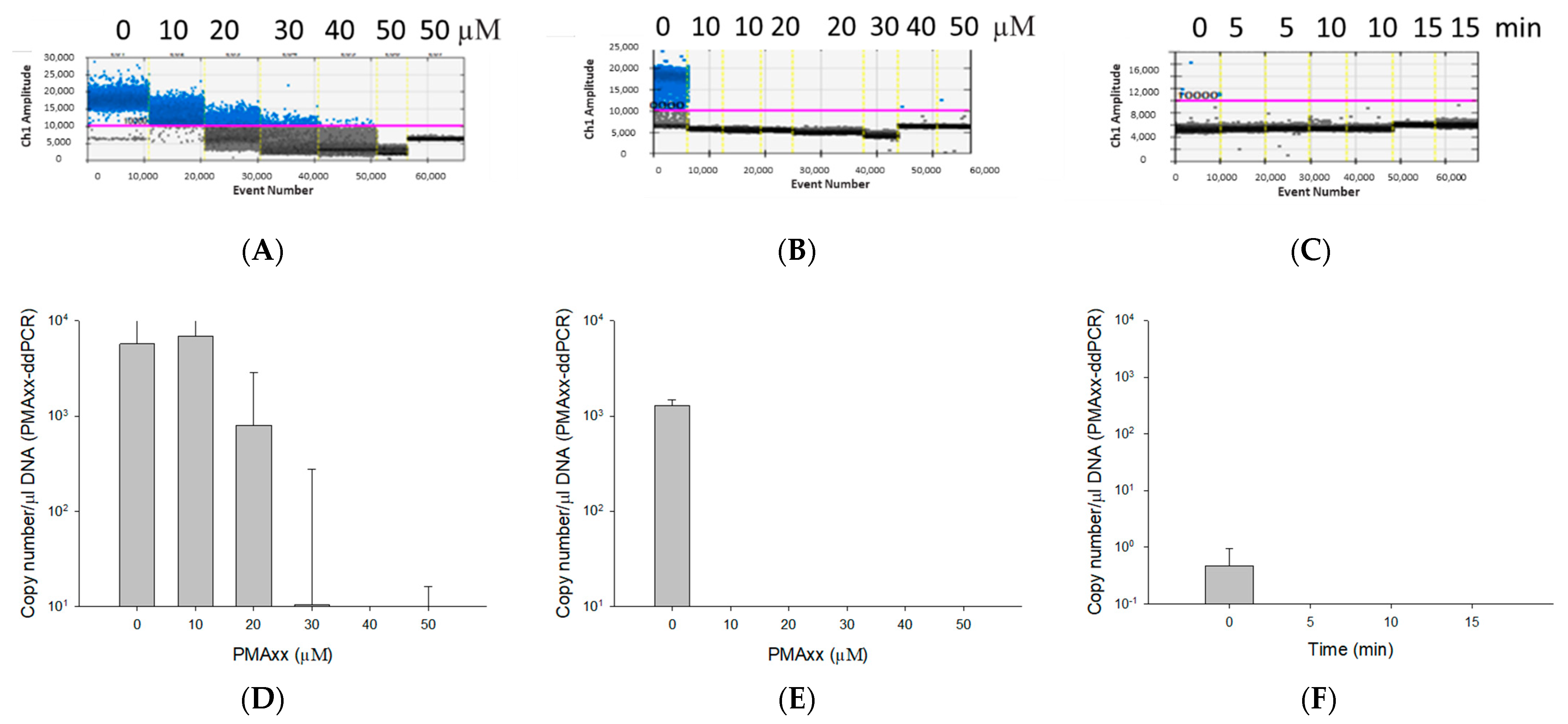
2.2. Optimization of PMAxx Treatment
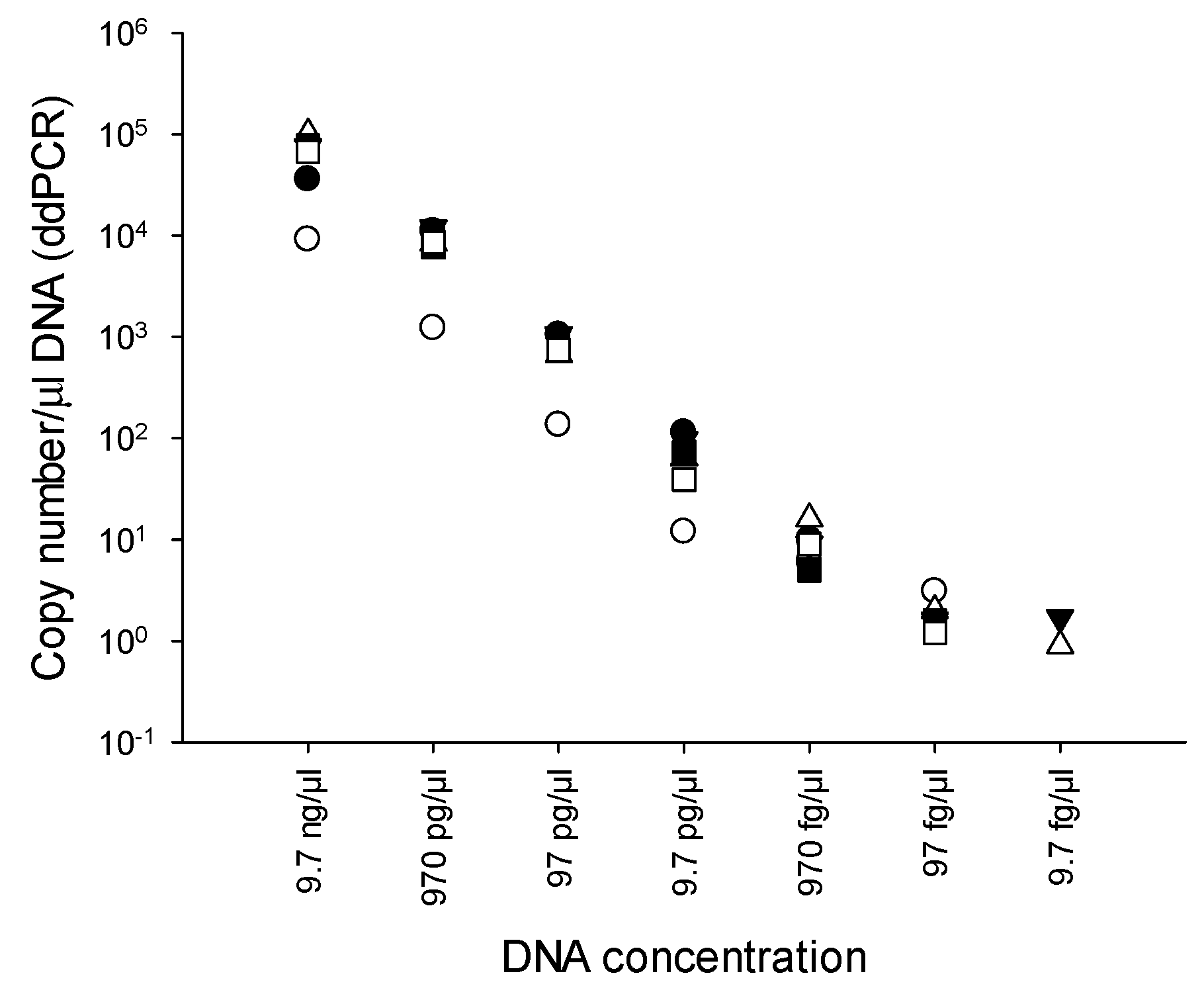
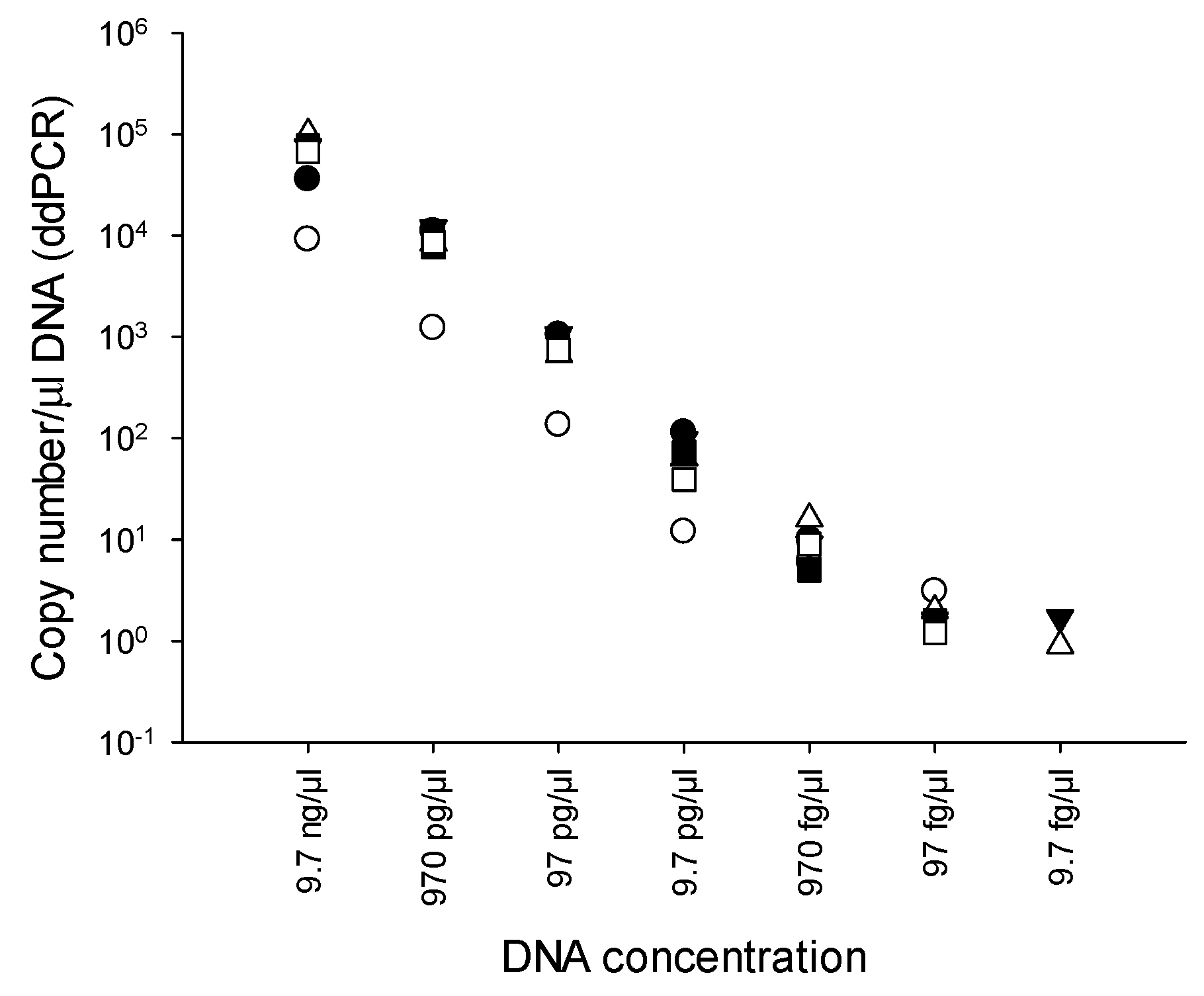
2.3. Limit of detection (LOD) of PMAxx-ddPCR
2.4. Assessment of the PMAxx-ddPCR Assay
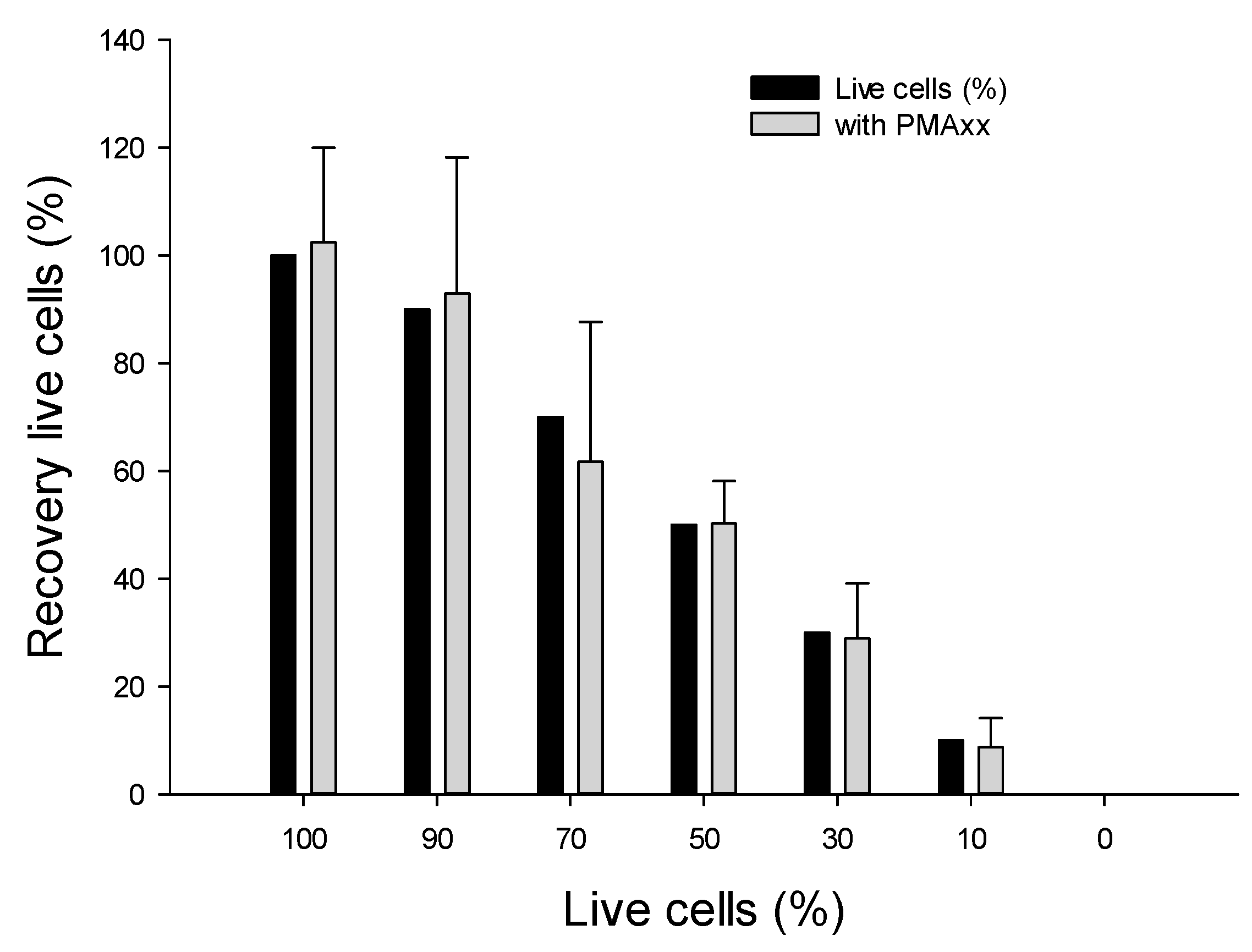
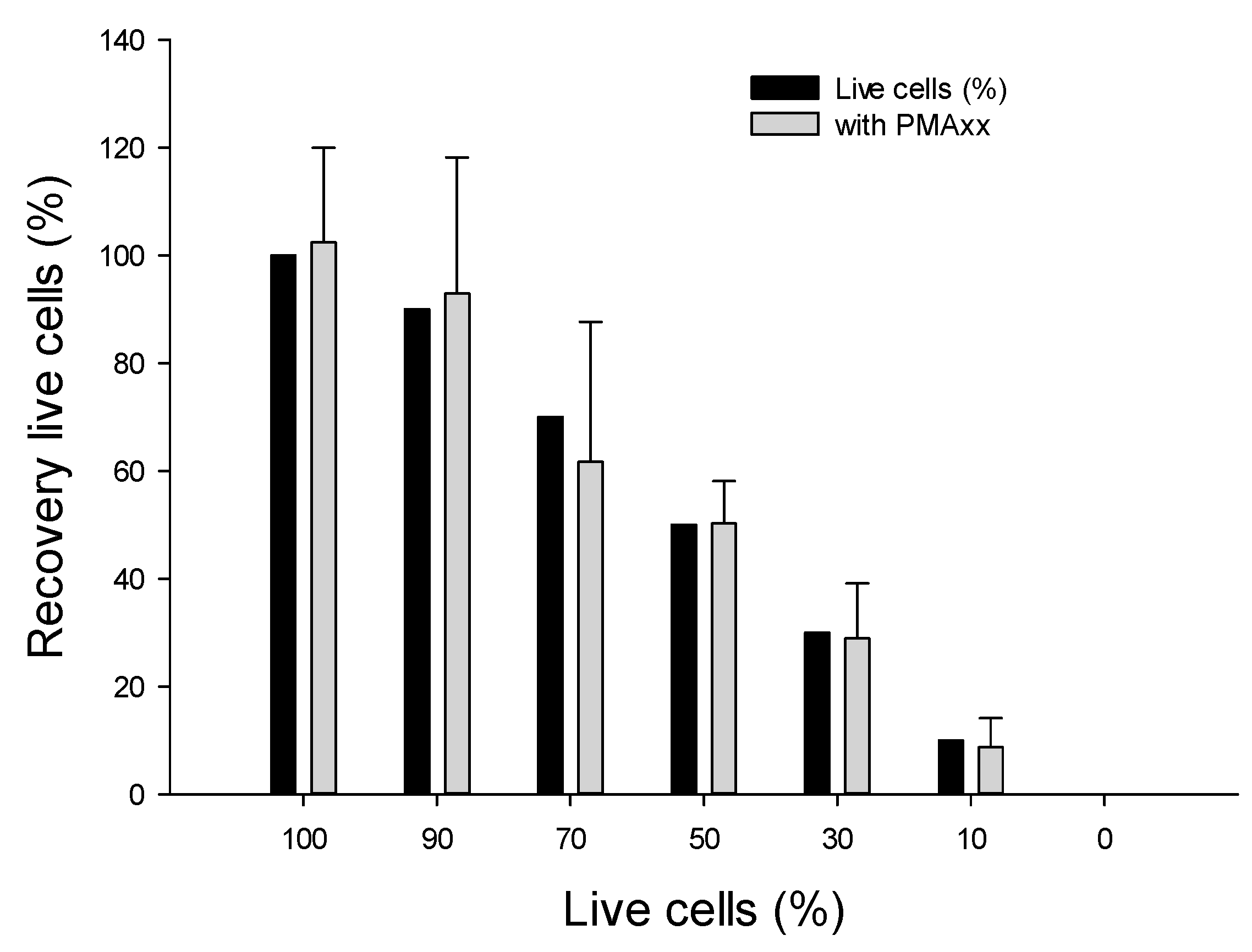
2.4.1. Using Various Ratios of Live vs. Dead B. cenocepacia J2315 Cells
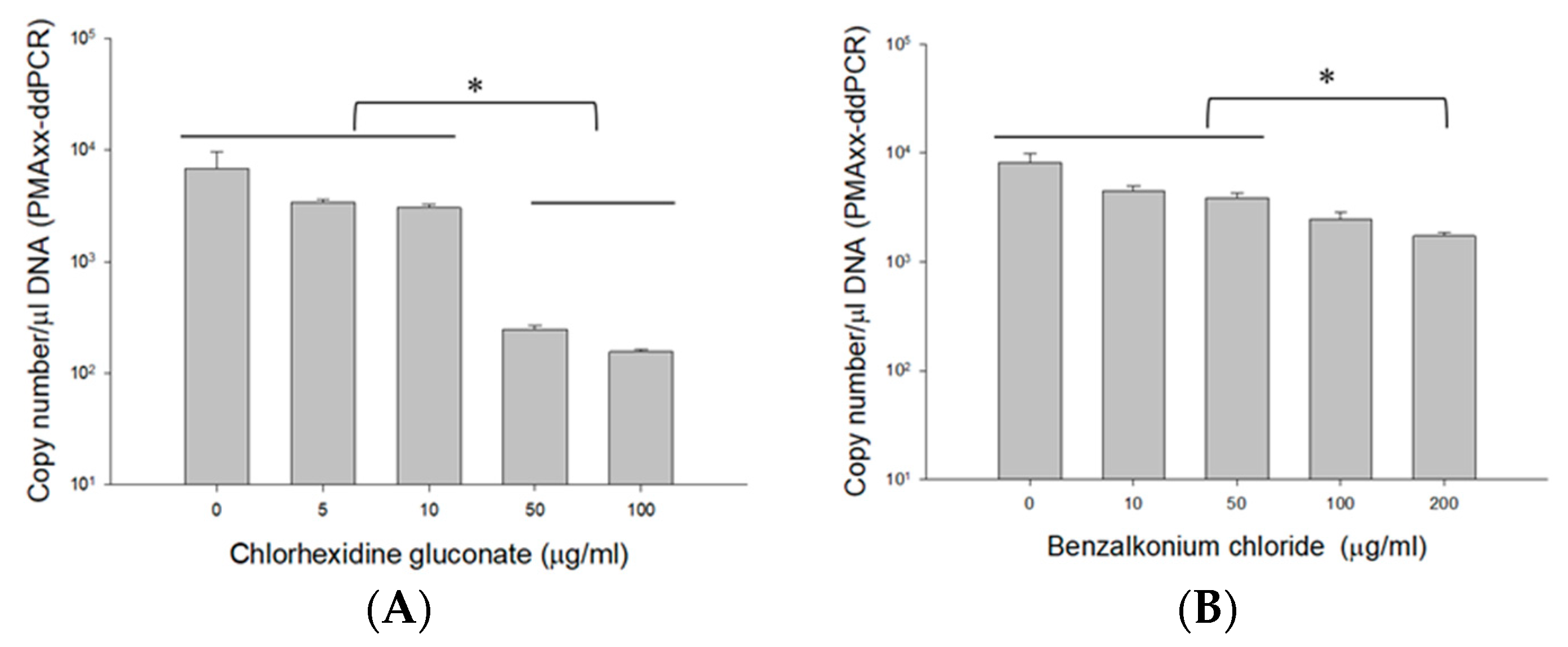
2.4.2. Using Different Concentrations of CHX and BZK
2.5. Application of the PMAxx ddPCR Assay with 20 BCC Strains in Nuclease-Free Water and Antiseptics
2.6. Statistical Analysis
3. Results
3.1. Evaluating the Primer Sets
3.2. Optimization of the PMAxx Concentration and Light Exposure Time
3.3. LOD of PMAxx-ddPCR
3.4. Assessment of the PMAxx-ddPCR Assay
3.4.1. Assessing Live/Dead Cells in Nuclease-Free Water
3.4.2. PMAxx-ddPCR Assay in CHX and BZK
3.5. Application of PMAxx-ddPCR Assay in Nuclease-Free Water and Antiseptics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Depoorter, E.; De Canck, E.; Peeters, C.; Wieme, A.D.; Cnockaert, M.; Zlosnik, J.E.A.; LiPuma, J.J.; Coenye, T.; Vandamme, P. Burkholderia cepacia complex taxon K: Where to split? Front. Microbiol. 2020, 11, 1594. [Google Scholar] [CrossRef] [PubMed]
- Tavares, M.; Kozak, M.; Balola, A.; Coutinho, C.P.; Godinho, C.P.; Hassan, A.A.; Cooper, V.S.; Sa-Correia, I. Adaptation and survival of Burkholderia cepacia and B. contaminans during long-term incubation in saline solutions containing benzalkonium chloride. Front. Bioeng. Biotechnol. 2020, 8, 630. [Google Scholar] [CrossRef] [PubMed]
- Mahenthiralingam, E.; Urban, T.A.; Goldberg, J.B. The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 2005, 3, 144–156. [Google Scholar] [CrossRef]
- LiPuma, J.J.; Dulaney, B.J.; McMenamin, J.D.; Whitby, P.W.; Stull, T.L.; Coenye, T.; Vandamme, P. Development of rRNA-based PCR assays for identification of Burkholderia cepacia complex isolates recovered from cystic fibrosis patients. J. Clin. Microbiol. 1999, 37, 3167–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, L. Microbial diversity in pharmaceutical product recalls and environments. PDA J. Pharm. Sci. Technol. 2007, 61, 383–399. [Google Scholar]
- Jimenez, L. Analysis of FDA enforcement reports (2012–2019) to determine the microbial diversity in contaminated non-sterile and sterile drugs. Am. Pharm. Rev. 2019, 4, 1–21. [Google Scholar]
- Sutton, S.; Jimenez, L. A review of reported recalls involving microbiological control 2004–2011 with emphasis on FDA considerations of “objectionable organisms”. Am. Pharm. Rev. 2012, 15, 42–57. [Google Scholar]
- Jimenez, L.; Jashari, T.; Vasquez, J.; Zapata, S.; Bochis, J.; Kulko, M.; Ellman, V.; Gardner, M.; Choe, T. Real-time PCR detection of Burkholderia cepacia in pharmaceutical products contaminated with low levels of bacterial contamination. PDA J. Pharm. Sci. Technol. 2018, 72, 73–80. [Google Scholar] [CrossRef]
- Ahn, Y.; Gibson, B.; Williams, A.; Alusta, P.; Buzatu, D.A.; Lee, Y.J.; LiPuma, J.J.; Hussong, D.; Marasa, B.; Cerniglia, C.E. A comparison of culture-based, real-time PCR, droplet digital PCR and flow cytometric methods for the detection of Burkholderia cepacia complex in nuclease-free water and antiseptics. J. Ind. Microbiol. Biotechnol. 2020, 47, 475–484. [Google Scholar] [CrossRef]
- Mahenthiralingam, E.; Bischof, J.; Byrne, S.K.; Radomski, C.; Davies, J.E.; Av-Gay, Y.; Vandamme, P. DNA-Based diagnostic approaches for identification of Burkholderia cepacia complex, Burkholderia vietnamiensis, Burkholderia multivorans, Burkholderia stabilis, and Burkholderia cepacia genomovars I and III. J. Clin. Microbiol. 2000, 38, 3165–3173. [Google Scholar] [CrossRef] [Green Version]
- Attia, M.A.; Ali, A.E.; Essam, T.M.; Amin, M.A. Direct detection of Burkholderia cepacia in susceptible pharmaceutical products using semi-nested PCR. PDA J. Pharm. Sci. Technol. 2016, 70, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreo, T.; Pirc, M.; Ramsak, Z.; Pavsic, J.; Milavec, M.; Zel, J.; Gruden, K. Optimising droplet digital PCR analysis approaches for detection and quantification of bacteria: A case study of fire blight and potato brown rot. Anal. Bioanal. Chem. 2014, 406, 6513–6528. [Google Scholar] [CrossRef]
- Emslie, K.R.; JL, H.M.; Griffiths, K.; Forbes-Smith, M.; Pinheiro, L.B.; Burke, D.G. Droplet volume variability and impact on digital PCR copy number concentration measurements. Anal. Chem. 2019, 91, 4124–4131. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Nshimyimana, J.P.; Cruz, M.C.; Wuertz, S.; Thompson, J.R. Variably improved microbial source tracking with digital droplet PCR. Water Res. 2019, 159, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Della Starza, I.; Nunes, V.; Cavalli, M.; De Novi, L.A.; Ilari, C.; Apicella, V.; Vitale, A.; Testi, A.M.; Del Giudice, I.; Chiaretti, S.; et al. Comparative analysis between RQ-PCR and digital-droplet-PCR of immunoglobulin/T-cell receptor gene rearrangements to monitor minimal residual disease in acute lymphoblastic leukaemia. Br. J. Haematol. 2016, 174, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Rudi, K.; Moen, B.; Dromtorp, S.M.; Holck, A.L. Use of ethidium monoazide and PCR in combination for quantification of viable and dead cells in complex samples. Appl. Environ. Microbiol. 2005, 71, 1018–1024. [Google Scholar] [CrossRef] [Green Version]
- Lazou, T.P.; Iossifidou, E.G.; Gelasakis, A.I.; Chaintoutis, S.C.; Dovas, C.I. Viability quantitative PCR Utilizing propidium monoazide, spheroplast formation, and Campylobacter coli as a bacterial model. Appl. Environ. Microbiol. 2019, 85, e01499-19. [Google Scholar] [CrossRef]
- Zhang, J.; Khan, S.; Chousalkar, K.K. Development of PMAxx(TM)-based qPCR for the quantification of viable and non-viable load of Salmonella from poultry environment. Front. Microbiol. 2020, 11, 581201. [Google Scholar] [CrossRef]
- Nocker, A.; Cheung, C.Y.; Camper, A.K. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J. Microbiol. Methods 2006, 67, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Wang, K.; Ma, L.; Delaquis, P.; Bach, S.; Feng, J.; Lu, X. Viable but nonculturable Escherichia coli O157:H7 and Salmonella enterica in fresh produce: Rapid determination by loop-mediated isothermal amplification coupled with a propidium monoazide treatment. Appl. Environ. Microbiol. 2020, 86, e02566-19. [Google Scholar] [CrossRef] [PubMed]
- Lazou, T.P.; Gelasakis, A.I.; Chaintoutis, S.C.; Iossifidou, E.G.; Dovas, C.I. Method-dependent implications in foodborne pathogen quantification: The case of Campylobacter coli survival on meat as comparatively assessed by colony count and viability PCR. Front. Microbiol. 2021, 12, 604933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Wang, L.L.; Lu, S.Y.; Hu, P.; Li, Y.S.; Zhang, Y.; Chang, H.Z.; Zhai, F.F.; Liu, Z.S.; Li, Z.H.; et al. A novel, rapid, and simple PMA-qPCR method for detection and counting of viable Brucella organisms. J. Vet. Res. 2020, 64, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Ferrando, E.; Randazzo, W.; Pérez-Cataluña, A.; Falcó, I.; Navarro, D.; Martin-Latil, S.; Díaz-Reolid, A.; Girón-Guzmán, I.; Allende, A.; Sanchez, G. Platinum chloride-based viability RT-qPCR for SARS-CoV-2 detection in complex samples. Sci. Rep. 2021, 11, 18120. [Google Scholar] [CrossRef]
- Randazzo, W.; Khezri, M.; Ollivier, J.; Le Guyader, F.S.; Rodriguez-Diaz, J.; Aznar, R.; Sanchez, G. Optimization of PMAxx pretreatment to distinguish between human norovirus with intact and altered capsids in shellfish and sewage samples. Int. J. Food Microbiol. 2018, 266, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Gu, X.; Wang, L.; He, X.; He, C.; Zhang, J.; Zhao, L. Rapid and absolute quantification of VBNC Cronobacter sakazakii by PMAxx combined with single intact cell droplet digital PCR in infant foods. LWT Food Sci. Technol. 2021, 145, 111388. [Google Scholar] [CrossRef]
- Lv, X.; Wang, L.; Zhang, J.; He, X.; Shi, L.; Zhao, L. Quantitative detection of trace VBNC Cronobacter sakazakii by immunomagnetic separation in combination with PMAxx-ddPCR in dairy products. Food Microbiol. 2021, 99, 103831. [Google Scholar] [CrossRef]
- Gobert, G.; Cotillard, A.; Fourmestraux, C.; Pruvost, L.; Miguet, J.; Boyer, M. Droplet digital PCR improves absolute quantification of viable lactic acid bacteria in faecal samples. J. Microbiol. Methods 2018, 148, 64–73. [Google Scholar] [CrossRef]
- Daddy Gaoh, S.; Kweon, O.; Lee, Y.-J.; LiPuma, J.J.; Hussong, D.; Marasa, B.; Ahn, Y. Loop-mediated isothermal amplification (LAMP) assay for detecting Burkholderia cepacia complex in non-sterile pharmaceutical products. Pathogens 2021, 10, 1071. [Google Scholar] [CrossRef]
- Wang, M.; Yang, J.; Gai, Z.; Huo, S.; Zhu, J.; Li, J.; Wang, R.; Xing, S.; Shi, G.; Shi, F.; et al. Comparison between digital PCR and real-time PCR in detection of Salmonella typhimurium in milk. Int. J. Food Microbiol. 2018, 266, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Harringer, M.; Alfreider, A. Primer evaluation and development of a droplet digital PCR protocol targeting amoA genes for the quantification of Comammox in lakes. Sci. Rep. 2021, 11, 2982. [Google Scholar] [CrossRef] [PubMed]
- Fittipaldi, M.; Nocker, A.; Codony, F. Progress in understanding preferential detection of live cells using viability dyes in combination with DNA amplification. J. Microbiol. Methods 2012, 91, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Soejima, T.; Iida, K.; Qin, T.; Taniai, H.; Seki, M.; Yoshida, S. Method to detect only live bacteria during PCR amplification. J. Clin. Microbiol. 2008, 46, 2305–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.M.; Ahn, Y.; LiPuma, J.J.; Hussong, D.; Cerniglia, C.E. Survival and susceptibility of Burkholderia cepacia complex in chlorhexidine gluconate and benzalkonium chloride. J. Ind. Microbiol. Biotechnol. 2015, 42, 905–913. [Google Scholar] [CrossRef]
- Ahn, Y.; Kim, J.M.; Kweon, O.; Kim, S.J.; Jones, R.C.; Woodling, K.; Gamboa da Costa, G.; LiPuma, J.J.; Hussong, D.; Marasa, B.S.; et al. Intrinsic resistance of Burkholderia cepacia complex to benzalkonium chloride. mBio 2016, 7, e01716-16. [Google Scholar] [CrossRef] [Green Version]
- Cangelosi, G.A.; Meschke, J.S. Dead or alive: Molecular assessment of microbial viability. Appl. Environ. Microbiol. 2014, 80, 5884–5891. [Google Scholar] [CrossRef] [Green Version]
- Cremonesi, P.; Cortimiglia, C.; Picozzi, C.; Minozzi, G.; Malvisi, M.; Luini, M.; Castiglioni, B. Development of a droplet digital polymerase chain reaction for rapid and simultaneous identification of common foodborne pathogens in soft cheese. Front. Microbiol. 2016, 7, 1725. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplicons Name | Primer Sequence (5—3) | Amplicon Size (bp) | |
|---|---|---|---|
| RibB67 | Forward Reverse | GCGATACGAAGGAACACCTG CGTAGCCGGACATGCTG | 189 |
| RibB5 | Forward Reverse | GGCCGGATGGTGATCCT GTCATCAGCGGCAGGTG | 176 |
| Tested Inoculum (CFU/mL) | Nuclease-Free Distilled Water | CHX | BZK | |||
|---|---|---|---|---|---|---|
| Without PMAxx | With PMAxx | Without PMAxx | With PMAxx | Without PMAxx | With PMAxx | |
| 104 | 50/60 a | 38/60 | 54/60 | 42/60 | 48/60 | 50/60 |
| 103 | 55/60 | 34/60 | 49/60 | 23/60 | 47/60 | 54/60 |
| 102 | 49/60 | 37/60 | 41/60 | 11/60 | 28/60 | 33/60 |
| 10 | 50/60 | 35/60 | 35/60 | 16/60 | 29/60 | 24/60 |
| 204/240 (85.0%) b | 144/240 (60.0%) | 179/240 (74.6%) | 92/240 (38.3%) | 152/240 (63.3%) | 161/240 (67.1%) | |
| p = 0.0163 c | p = 0.0001 | p = 0.7166 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daddy Gaoh, S.; Kweon, O.; Lee, Y.-J.; Hussong, D.; Marasa, B.; Ahn, Y. A Propidium Monoazide (PMAxx)-Droplet Digital PCR (ddPCR) for the Detection of Viable Burkholderia cepacia Complex in Nuclease-Free Water and Antiseptics. Microorganisms 2022, 10, 943. https://doi.org/10.3390/microorganisms10050943
Daddy Gaoh S, Kweon O, Lee Y-J, Hussong D, Marasa B, Ahn Y. A Propidium Monoazide (PMAxx)-Droplet Digital PCR (ddPCR) for the Detection of Viable Burkholderia cepacia Complex in Nuclease-Free Water and Antiseptics. Microorganisms. 2022; 10(5):943. https://doi.org/10.3390/microorganisms10050943
Chicago/Turabian StyleDaddy Gaoh, Soumana, Ohgew Kweon, Yong-Jin Lee, David Hussong, Bernard Marasa, and Youngbeom Ahn. 2022. "A Propidium Monoazide (PMAxx)-Droplet Digital PCR (ddPCR) for the Detection of Viable Burkholderia cepacia Complex in Nuclease-Free Water and Antiseptics" Microorganisms 10, no. 5: 943. https://doi.org/10.3390/microorganisms10050943
APA StyleDaddy Gaoh, S., Kweon, O., Lee, Y.-J., Hussong, D., Marasa, B., & Ahn, Y. (2022). A Propidium Monoazide (PMAxx)-Droplet Digital PCR (ddPCR) for the Detection of Viable Burkholderia cepacia Complex in Nuclease-Free Water and Antiseptics. Microorganisms, 10(5), 943. https://doi.org/10.3390/microorganisms10050943