
Easy Express Extraction (TripleE)—A Universal, Electricity-Free Nucleic Acid Extraction System for the Lab and the Pen


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Viruses
2.2. Nucleic Acid Extraction
2.2.1. KingFisher Flex Extraction System
2.2.2. IndiMag 48 Extraction System
2.2.3. Easy Express Extraction (TripleE) System
Extraction Instrument
Extraction Plate and Buffers
Extraction Workflow
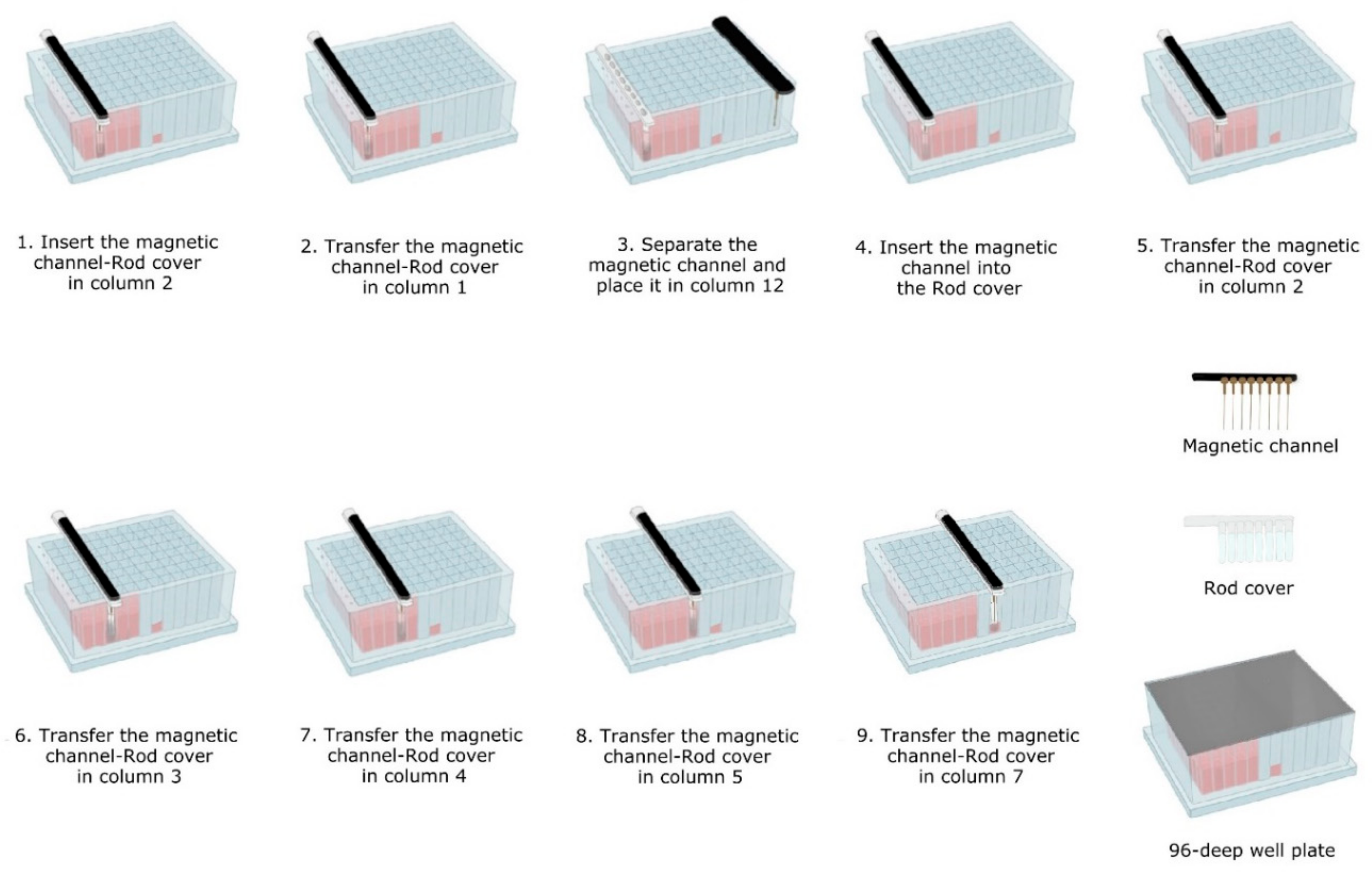
- Lysis-binding steps: A 100 µL sample was placed on 1.5 mL tubes (Eppendorf) prefilled with 100 µL VXL lysis buffer and 400 µL ACB binding buffer (IndiMag Pathogen Kit, Indical Bioscience). Then the 600 µL sample-lysis-binding mix was thoroughly mixed by repeated pipetting and added to the first column (including the Proteinase K) of the prefilled 96-deep well plate.Then, magnetic beads were collected from column 2 with the magnetic channel inserted into the rod cover. This was carried out by dipping up and down the magnetic channel-rod cover up to 10 times (Figure 3—1). Subsequently, the magnetic channel-rod cover with the attached magnetic beads was transferred into column 1 (Figure 3—2), then the magnetic channel was removed and placed the parking position in column 12. Now, the separate rod cover in column 1 was dipped up and down 30 times and was incubated for 3 min at RT (Figure 3—3). Next, the magnetic channel, picked up from the park position, was inserted into the rod cover (Figure 3—4), and the combo was dipped slowly 10 times up and down to collect the magnetic beads again.
- Washing steps: The magnetic channel-rod cover with the attached magnetic beads was inserted into column 2 (Figure 3—5). The washing step was performed by dipping up and down 30 times with the combined magnetic channel-rod cover without the complete releasing of the magnetic beads. Detached beads were recollected by dipping with slower movements 10 times. This latter step was used to catch the maximum number of magnetic beads free in solution. Subsequently, the described washing procedure was applied to the next three washing steps using columns 3, 4 and 5 (Figure 3—6–8).
- Elution step: Finally, the magnetic channel-rod cover with the attached magnetic beads was inserted into column 7 (Figure 3—9) and was dipped up and down 30 times, again followed by a dipping step consisting of 10 slower movements for catching the maximum number of magnetic beads. Thereafter, the rod cover and the attached magnetic beads were discarded. The ready-to-use nucleic acids remained in column 7 for subsequent real-time PCR amplification or other molecular analyses.
2.3. Real-Time PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCR Assay | Genome Detection of | Primer/Probe | Sequence 5′-3′ | Amplicon (Base Pair) | Reference |
|---|---|---|---|---|---|
| ASFV-P72-IVI-mix | ASFV | ASFV-p72IVI-F | GAT GAT GAT TAC CTT YGC TTT GAA | 78 | Haines et al., 2013 [30] |
| ASFV-p72IVI-R | TCT CTT GCT CTR GAT ACR TTA ATA TGA | ||||
| ASFV-p72IVI-FAM | FAM-CCA CGG GAG GAA TAC CAA CCC AGT G-BHQ1 | ||||
| Capri-p32-mix | Capripoxvirus | Capri-p32for | AAA ACG GTA TAT GGA ATA GAG TTG GAA | 89 | Bowden et al., 2008 [31] modified; Dietze et al., 2018 [32] |
| Capri-p32rev | AAA TGA AAC CAA TGG ATG GGA TA | ||||
| Capri-p32-FAM | FAM-ATG GAT GGC TCA TAG ATT TCC TGA T-BHQ1 | ||||
| Pan BTV-IVI-mix | BTV | Orru_BTV_IVI_F2 | TGG AYA AAG CRA TGT CAA A | 97 | OIE terrestrial manual (version May 2021) |
| Orru_BTV_IVI_R2 | ACR TCA TCA CGA AAC GCT TC | ||||
| Orru_BTV_IVI_FAM | FAM-ARG CTG CAT TCG CAT CGT ACG C-BHQ1 | ||||
| PPRV-Batten-mix | PPRV | PPRV-N-483F | AGA GTT CAA TAT GTT RTT AGC CTC CAT | 142 | Batten et al., 2011 [34] |
| PPRV-N-624R | TTC CCC ART CAC TCT YCT TTG T | ||||
| PPRV-N-551FAM | FAM-CAC CGG AYA CKG CAG CTG ACT CAG AA-BHQ1 | ||||
| ß-Actin-DNA-mix 2 | beta-actin mRNA | ACT-1030-F | AGC GCA AGT ACT CCG TGT G | 106 | Toussaint et al., 2007 [35] modified; Wernike et al., 2011 [36] |
| ACT-1135-R | CGG ACT CAT CGT ACT CCT GCT T | ||||
| ACT-1081-HEX | HEX-TCG CTG TCC ACC TTC CAG CAG ATG T-BHQ1 |
2.4. Data Analyses and Statistics
3. Results
3.1. Reproducibility of the Extraction Methods
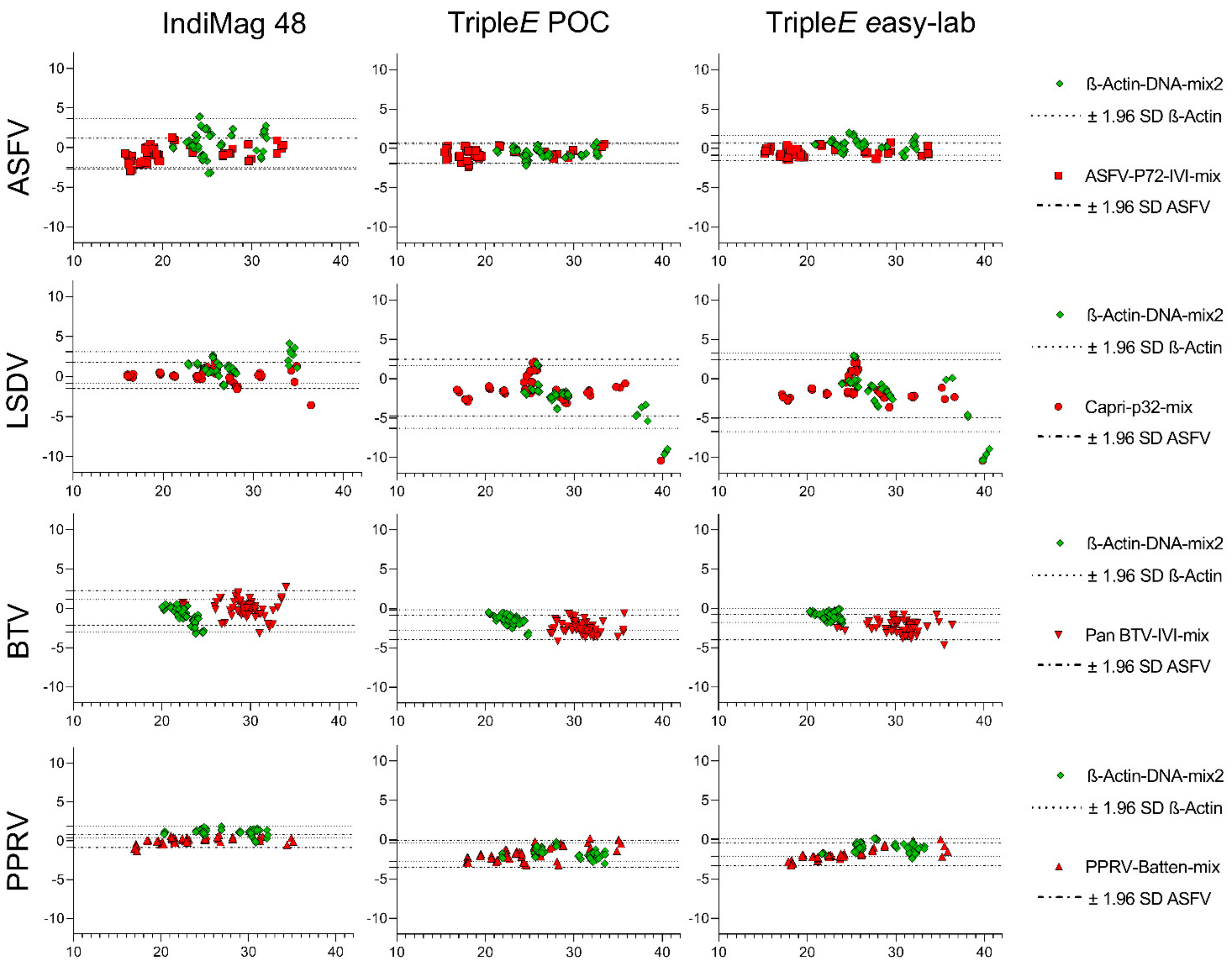
3.2. Analytical Performance of the Extraction Methods
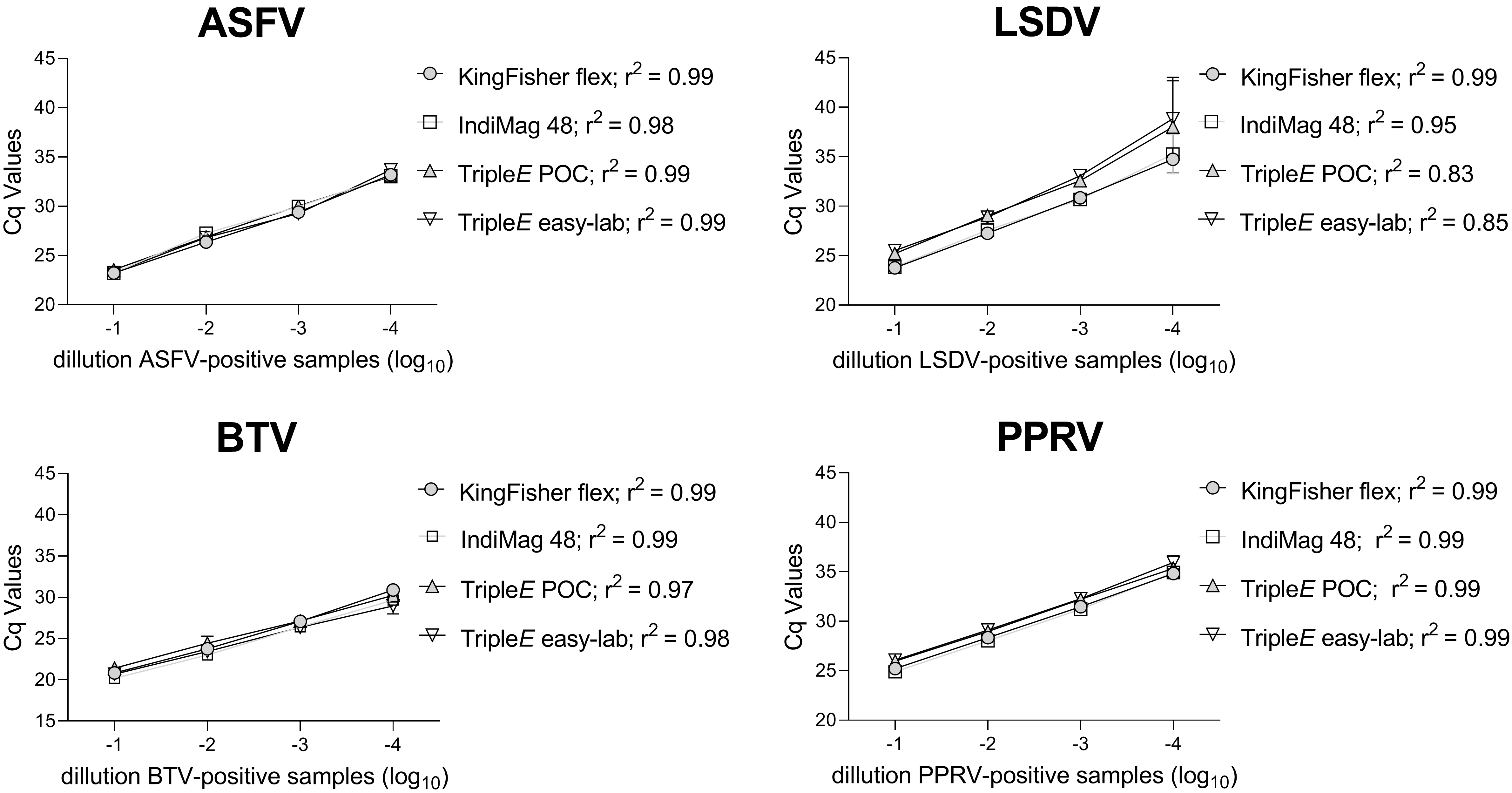
3.3. Linearity and Analytical Sensitivity of Extraction Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Babiuk, S.; Bowden, T.R.; Boyle, D.B.; Wallace, D.B.; Kitching, R.P. Capripoxviruses: An Emerging Worldwide Threat to Sheep, Goats and Cattle. Transbound. Emerg. Dis. 2008, 55, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, V.; Hemadri, D.; Gajendragad, M.R.; Singh, R.K.; Rahman, H. Diagnosis and control of peste des petits ruminants: A comprehensive review. VirusDis. 2013, 25, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Rajko-Nenow, P.; Christodoulou, V.; Thurston, W.; Ropiak, H.M.; Savva, S.; Brown, H.; Qureshi, M.; Alvanitopoulos, K.; Gubbins, S.; Flannery, J.; et al. Origin of Bluetongue Virus Serotype 8 Outbreak in Cyprus, September 2016. Viruses 2020, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Landeg, F. Bluetongue outbreak in the UK. Vet. Rec. 2007, 161, 534. [Google Scholar] [CrossRef]
- Kyriakis, C.S.; Billinis, C.; Papadopoulos, E.; Vasileiou, N.G.; Athanasiou, L.V.; Fthenakis, G.C. Bluetongue in small ruminants: An opinionated review, with a brief appraisal of the 2014 outbreak of the disease in Greece and the south-east Europe. Vet. Microbiol. 2015, 181, 66–74. [Google Scholar] [CrossRef]
- Blome, S.; Franzke, K.; Beer, M. African swine fever—A review of current knowledge. Virus Res. 2020, 287, 198099. [Google Scholar] [CrossRef]
- Pikalo, J.; Schoder, M.E.; Sehl, J.; Breithaupt, A.; Tignon, M.; Cay, A.B.; Gager, A.M.; Fischer, M.; Beer, M.; Blome, S. The African swine fever virus isolate Belgium 2018/1 shows high virulence in European wild boar. Transbound. Emerg. Dis. 2020, 67, 1654–1659. [Google Scholar] [CrossRef]
- Sauter-Louis, C.; Schulz, K.; Richter, M.; Staubach, C.; Mettenleiter, T.C.; Conraths, F.J. African swine fever: Why the situation in Germany is not comparable to that in the Czech Republic or Belgium. Transbound. Emerg. Dis. 2021. [Google Scholar] [CrossRef]
- Aebischer, A.; Beer, M.; Hoffmann, B. Development and validation of rapid magnetic particle based extraction protocols. Virol. J. 2014, 11, 137. [Google Scholar] [CrossRef]
- OIE-World organisation for animal Health. Manual of diagnostic tests and vaccines for terrestrial animals. In Development and Optimisation of Nucleic Acid Detection Assays; OIE: Paris, France, 2021; Chapter 2.2.3; Available online: https://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.02.03_NAD_ASSAYS.pdf (accessed on 1 July 2018).
- Kim, S.; Kim, J.H.; Kim, S.; Park, J.S.; Cha, B.S.; Lee, E.S.; Han, J.; Shin, J.; Jang, Y.; Park, K.S. Loop-mediated isothermal amplification-based nucleic acid lateral flow assay for the specific and multiplex detection of genetic markers. Anal. Chim. Acta 2022, 1205, 339781. [Google Scholar] [CrossRef]
- Yoon, T.; Shin, J.; Choi, H.J.; Park, K.S. Split T7 promoter-based isothermal transcription amplification for one-step fluorescence detection of SARS-CoV-2 and emerging variants. Biosens. Bioelectron. 2022, 208, 114221. [Google Scholar] [CrossRef] [PubMed]
- Ballagi-Pordány, A.; Belák, S. The use of mimics as internal standards to avoid false negatives in diagnostic PCR. Mol. Cell. Probes 1996, 10, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Belák, S.; Thorén, P. Molecular diagnosis of animal diseases: Some experiences over the past decade. Expert Rev. Mol. Diagn. 2001, 1, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Belák, S. The molecular diagnosis of porcine viral diseases: A review. Acta Vet. Hung. 2005, 53, 113–124. [Google Scholar] [CrossRef][Green Version]
- Belák, S. Molecular diagnosis of viral diseases, present trends and future aspects: A view from the OIE Collaborating Centre for the Application of Polymerase Chain Reaction Methods for Diagnosis of Viral Diseases in Veterinary Medicine. Vaccine 2007, 25, 5444–5452. [Google Scholar] [CrossRef]
- Burns, M.J.; Nixon, G.J.; A Foy, C.; Harris, N. Standardisation of data from real-time quantitative PCR methods—Evaluation of outliers and comparison of calibration curves. BMC Biotechnol. 2005, 5, 31. [Google Scholar] [CrossRef]
- Bustin, S.A. Real-time, fluorescence-based quantitative PCR: A snapshot of current procedures and preferences. Expert Rev. Mol. Diagn. 2005, 5, 493–498. [Google Scholar] [CrossRef]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6, 279–284. [Google Scholar] [CrossRef]
- Lauerman, L.H. Advances in PCR technology. Anim. Health Res. Rev. 2004, 5, 247–248. [Google Scholar] [CrossRef]
- Louie, M.; Louie, L.; Simor, A.E. The role of DNA amplification technology in the diagnosis of infectious diseases. CMAJ 2000, 163, 301–309. [Google Scholar]
- Endres, H.N.; Johnson, J.A.C.; Ross, C.A.; Welp, J.K.; Etzel, M.R. Evaluation of an ion-exchange membrane for the purification of plasmid DNA. Biotechnol. Appl. Biochem. 2003, 37, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, S.A. DNA/RNA Preparation for Molecular Detection. Clin. Chem. 2015, 61, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Knepp, J.H.; Geahr, M.A.; Forman, M.S.; Valsamakis, A. Comparison of Automated and Manual Nucleic Acid Extraction Methods for Detection of Enterovirus RNA. J. Clin. Microbiol. 2003, 41, 3532–3536. [Google Scholar] [CrossRef] [PubMed]
- Schlottau, K.; Freuling, C.M.; Müller, T.; Beer, M.; Hoffmann, B. Development of molecular confirmation tools for swift and easy rabies diagnostics. Virol. J. 2017, 14, 184. [Google Scholar] [PubMed]
- Shin, J.; Yoon, T.; Park, J.; Park, K.S. Sensitive and simultaneous detection of hygiene indicator bacteria using an enhanced CRISPR/Cas system in combination with a portable fluorescence detector. Sens. Actuators B Chem. 2022, 365, 131871. [Google Scholar] [CrossRef]
- Yoon, T.; Kim, S.; Kim, J.; Park, K. A Syringe-Based and Centrifugation-Free DNA Extraction Procedure for the Rapid Detection of Bacteria. Chemosensors 2021, 9, 167. [Google Scholar] [CrossRef]
- Elnagar, A.; Pikalo, J.; Beer, M.; Blome, S.; Hoffmann, B. Swift and Reliable “Easy Lab” Methods for the Sensitive Molecular Detection of African Swine Fever Virus. Int. J. Mol. Sci. 2021, 22, 2307. [Google Scholar] [CrossRef]
- Haines, F.J.; Hofmann, M.A.; King, D.P.; Drew, T.W.; Crooke, H.R. Development and validation of a multiplex, real-time RT PCR assay for the simultaneous detection of classical and African swine fever viruses. PLoS ONE 2013, 8, e71019. [Google Scholar] [CrossRef]
- Bowden, T.R.; Babiuk, S.L.; Parkyn, G.R.; Copps, J.S.; Boyle, D.B. Capripoxvirus tissue tropism and shedding: A quantitative study in experimentally infected sheep and goats. Virology 2008, 371, 380–393. [Google Scholar] [CrossRef]
- Dietze, K.; Moritz, T.; Alexandrov, T.; Krstevski, K.; Schlottau, K.; Milovanovic, M.; Hoffmann, D.; Hoffmann, B. Suitability of group-level oral fluid sampling in ruminant populations for lumpy skin disease virus detection. Vet. Microbiol. 2018, 221, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Eschbaumer, M.; Beer, M. Real-Time Quantitative Reverse Transcription-PCR Assays Specifically Detecting Bluetongue Virus Serotypes 1, 6, and 8. J. Clin. Microbiol. 2009, 47, 2992–2994. [Google Scholar] [CrossRef] [PubMed]
- Batten, C.A.; Banyard, A.C.; King, D.P.; Henstock, M.R.; Edwards, L.; Sanders, A.; Buczkowski, H.; Oura, C.C.; Barrett, T. A real time RT-PCR assay for the specific detection of Peste des petits ruminants virus. J. Virol. Methods 2011, 171, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, J.F.; Sailleau, C.; Breard, E.; Zientara, S.; De Clercq, K. Bluetongue virus detection by two real-time RT-qPCRs targeting two different genomic segments. J. Virol. Methods 2007, 140, 115–123. [Google Scholar] [CrossRef]
- Wernike, K.; Hoffmann, B.; Kalthoff, D.; König, P.; Beer, M. Development and validation of a triplex real-time PCR assay for the rapid detection and differentiation of wild-type and glycoprotein E-deleted vaccine strains of Bovine herpesvirus type 1. J. Virol. Methods 2011, 174, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Bland, J.M.; Altman, D.G. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1986, 1, 307–310. [Google Scholar] [CrossRef]
- Wang, T.; Sun, Y.; Qiu, H.J. African swine fever: An unprecedented disaster and challenge to China. Infect. Dis. Poverty 2018, 7, 111. [Google Scholar] [CrossRef]
- Parida, S.; Muniraju, M.; Altan, E.; Baazizi, R.; Raj, G.D.; Mahapatra, M. Emergence of PPR and its threat to Europe. Small Rumin. Res. 2016, 142, 16–21. [Google Scholar] [CrossRef]
- Pikalo, J.; Carrau, T.; Deutschmann, P.; Fischer, M.; Schlottau, K.; Beer, M.; Blome, S. Performance Characteristics of Real-Time PCRs for African Swine Fever Virus Genome Detection—Comparison of Twelve Kits to an OIE-Recommended Method. Viruses 2022, 14, 220. [Google Scholar] [CrossRef]
- Halecker, S.; Mettenleiter, T.C.; Beer, M.; Hoffmann, B. “FastCheck(FLI) PPR-like”—A Molecular Tool for the Fast Genome Detection of PPRV and Differential Diagnostic Pathogens. Viruses 2020, 12, 1227. [Google Scholar] [CrossRef]
- Voelker, C.R.; Ochoa, A.R.; Lott, L.; McDaniel, J.S.; Blackburn, A.N.; Cornell, L.E.; Mahoney, R.; Asin, S.N. Evaluating sensitivity and specificity of the Biomeme Franklin™ three9 real-time PCR device and SARS-CoV-2 go-strips assay using clinical samples. J. Clin. Virol. 2022, 146, 105046. [Google Scholar] [CrossRef] [PubMed]
- Hole, K.; Nfon, C. Foot-and-mouth disease virus detection on a handheld real-time polymerase chain reaction platform. Transbound. Emerg. Dis. 2019, 66, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Ambagala, A.; Fisher, M.; Goolia, M.; Nfon, C.; Furukawa-Stoffer, T.; Ortega Polo, R.; Lung, O. Field-Deployable Reverse Transcription-Insulated Isothermal PCR (RT-iiPCR) Assay for Rapid and Sensitive Detection of Foot-and-Mouth Disease Virus. Transbound. Emerg. Dis. 2017, 64, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Howson, E.L.A.; Armson, B.; Lyons, N.; Chepkwony, E.; Kasanga, C.J.; Kandusi, S.; Ndusilo, N.; Yamazaki, W.; Gizaw, D.; Cleaveland, S.; et al. Direct detection and characterization of foot-and-mouth disease virus in East Africa using a field-ready real-time PCR platform. Transbound. Emerg. Dis. 2018, 65, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Till, B.J.; Jankowicz-Cieslak, J.; Huynh, O.A.; Beshir, M.M.; Laport, R.G.; Hofinger, B.J. Low-Cost DNA Extraction. In Low-Cost Methods for Molecular Characterization of Mutant Plants: Tissue Desiccation, DNA Extraction and Mutation Discovery: Protocols; Springer: Cham, Switzerland, 2015; pp. 13–17. [Google Scholar]
- Möller, J.; Moritz, T.; Schlottau, K.; Krstevski, K.; Hoffmann, D.; Beer, M.; Hoffmann, B. Experimental lumpy skin disease virus infection of cattle: Comparison of a field strain and a vaccine strain. Arch. Virol. 2019, 164, 2931–2941. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, L.; Noll, L.; Stoy, C.; Porter, E.; Fu, J.; Feng, Y.; Peddireddi, L.; Liu, X.; Dodd, K.A.; et al. Development of a real-time PCR assay for detection of African swine fever virus with an endogenous internal control. Transbound. Emerg. Dis. 2020, 67, 2446–2454. [Google Scholar] [CrossRef] [PubMed]





| Virus | Extraction | Mean Ct | SD | CV (%) |
|---|---|---|---|---|
| ASFV | KingFisher Flex | 21.03 | 0.20 | 0.92 |
| IndiMag 48 | 21.77 | 0.26 | 1.18 | |
| TripleE POC | 21.63 | 0.18 | 0.93 | |
| TripleE easy-lab | 21.47 | 0.17 | 0.81 | |
| LSDV | KingFisher Flex | 24.87 | 0.16 | 0.63 |
| IndiMag 48 | 24.72 | 0.27 | 0.98 | |
| TripleE POC | 25.82 | 0.18 | 0.01 | |
| TripleE easy-lab | 25.99 | 0.20 | 0.72 | |
| PPRV | KingFisher Flex | 23.87 | 0.14 | 0.59 |
| IndiMag 48 | 23.44 | 0.18 | 0.77 | |
| TripleE POC | 25.23 | 0.30 | 1.13 | |
| TripleE easy-lab | 25.28 | 0.15 | 0.56 | |
| BTV | KingFisher Flex | 29.14 | 0.33 | 1.12 |
| IndiMag 48 | 29.07 | 0.50 | 1.72 | |
| TripleE POC | 31.55 | 0.41 | 1.29 | |
| TripleE easy-lab | 31.51 | 0.51 | 1.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korthase, C.; Elnagar, A.; Beer, M.; Hoffmann, B. Easy Express Extraction (TripleE)—A Universal, Electricity-Free Nucleic Acid Extraction System for the Lab and the Pen. Microorganisms 2022, 10, 1074. https://doi.org/10.3390/microorganisms10051074
Korthase C, Elnagar A, Beer M, Hoffmann B. Easy Express Extraction (TripleE)—A Universal, Electricity-Free Nucleic Acid Extraction System for the Lab and the Pen. Microorganisms. 2022; 10(5):1074. https://doi.org/10.3390/microorganisms10051074
Chicago/Turabian StyleKorthase, Christian, Ahmed Elnagar, Martin Beer, and Bernd Hoffmann. 2022. "Easy Express Extraction (TripleE)—A Universal, Electricity-Free Nucleic Acid Extraction System for the Lab and the Pen" Microorganisms 10, no. 5: 1074. https://doi.org/10.3390/microorganisms10051074
APA StyleKorthase, C., Elnagar, A., Beer, M., & Hoffmann, B. (2022). Easy Express Extraction (TripleE)—A Universal, Electricity-Free Nucleic Acid Extraction System for the Lab and the Pen. Microorganisms, 10(5), 1074. https://doi.org/10.3390/microorganisms10051074