
Composition, Structure and Diversity of Soil Bacterial Communities before, during and after Transit through the Gut of the Earthworm Aporrectodea caliginosa



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Soil, Earthworm Gut and Cast Sampling
2.2. Amplification, Sequencing and Analysis of 16S rRNA Genes
2.3. Statistical Analysis
3. Results
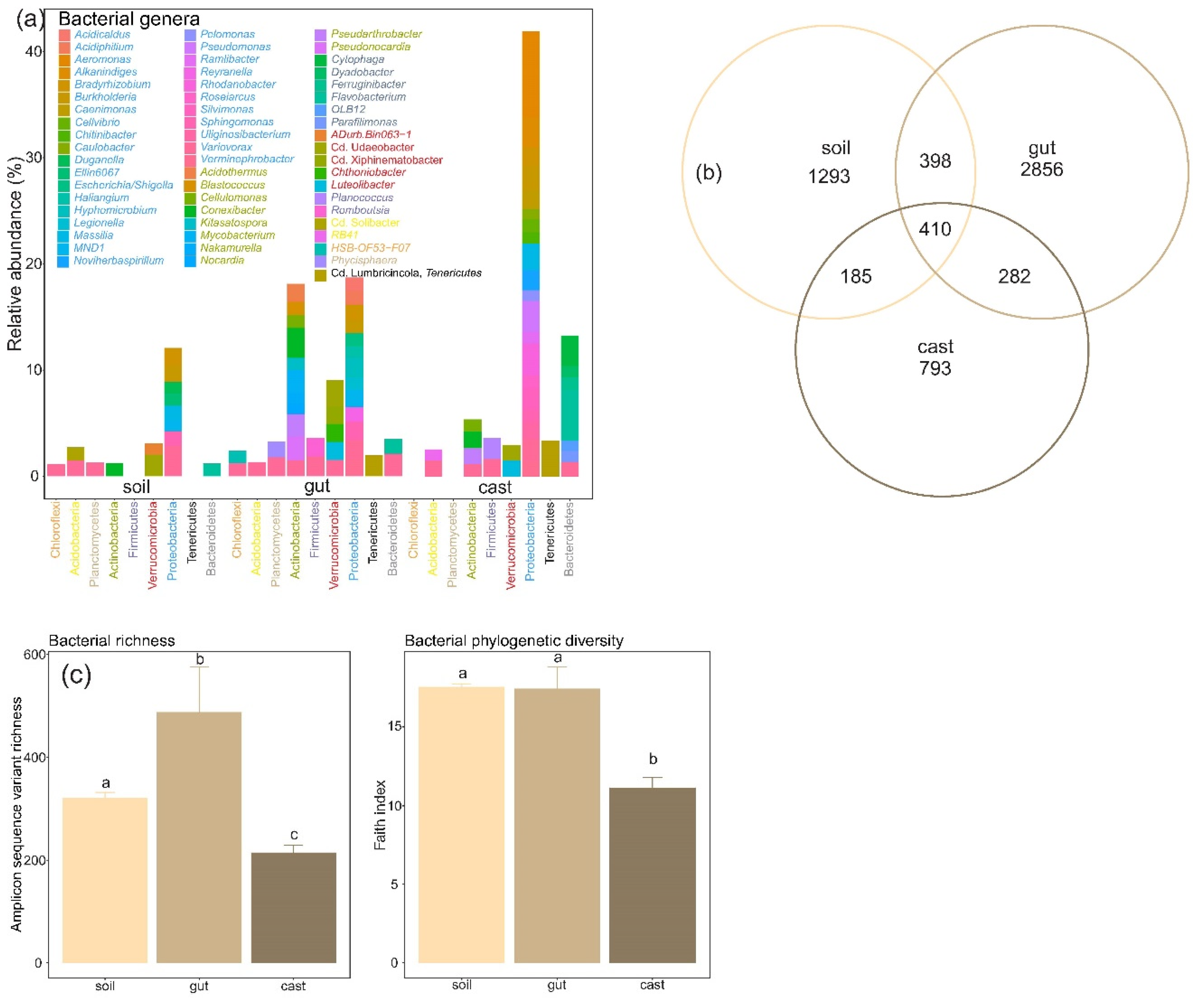
3.1. Composition of Soil and Earthworm Gut and Cast Microbiomes
3.2. Structure and Diversity of Soil and Earthworm Gut and Cast Microbiomes
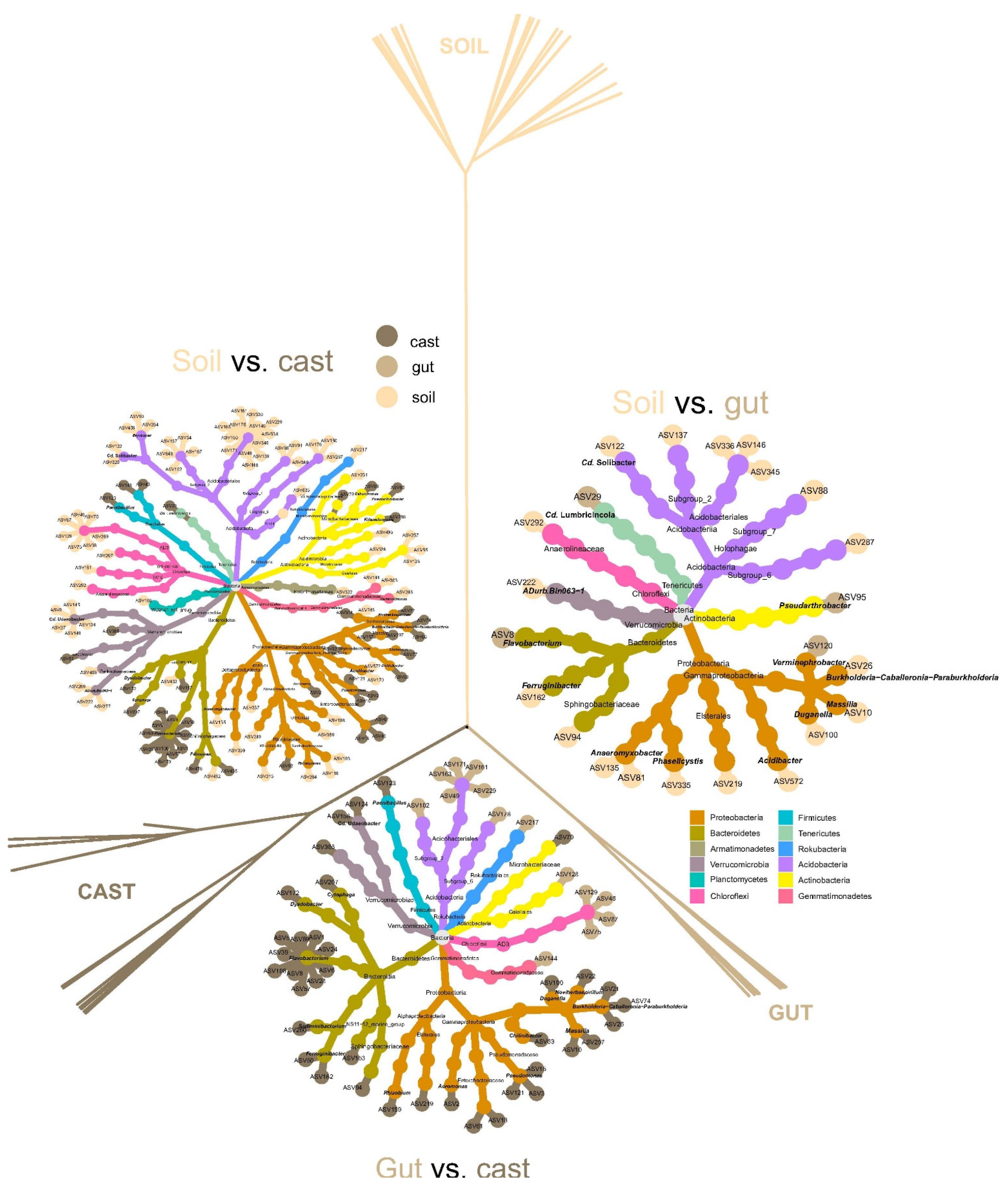
3.3. Metacommunity Assembly of Soil and Earthworm Gut and Cast Microbiomes
4. Discussion
4.1. Composition of Soil and Earthworm Gut and Cast Microbiomes
4.2. Structure and Diversity of Soil and Earthworm Gut and Cast Microbiomes
4.3. Metacommunity Assembly of Soil and Earthworm Gut and Cast Microbiomes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aira, M.; Domínguez, J. Changes in nutrient pools, microbial biomass and microbial activity in soils after transit through the gut of three endogeic earthworm species of the genus Postandrilus Qui and Bouché, 1998. J. Soil. Sediments 2014, 14, 1335–1340. [Google Scholar] [CrossRef]
- Aira, M.; Monroy, F.; Domínguez, J. Effects of two species of earthworms (Allolobophora spp.) on soil systems: A microfaunal and biochemical analysis. Pedobiologia 2003, 47, 877–881. [Google Scholar] [CrossRef][Green Version]
- Aira, M.; Sampedro, L.; Monroy, F.; Domínguez, J. Detritivorous earthworms directly modify the structure, thus altering the functioning of a microdecomposer food web. Soil Biol. Biochem. 2008, 40, 2511–2516. [Google Scholar] [CrossRef]
- Aira, M.; McNamara, N.; Piearce, T.; Domínguez, J. Microbial communities of Lumbricus terrestris (L.) middens: Structure, activity and changes through time in relation to earthworm presence. J. Soil. Sediments 2009, 9, 54–61. [Google Scholar] [CrossRef]
- Gómez-Brandón, M.; Lazcano, C.; Lores, M.; Domínguez, J. Detritivorous earthworms modify microbial community structure and accelerate plant residue decomposition. Appl. Soil Ecol. 2010, 44, 237–244. [Google Scholar] [CrossRef]
- Drake, H.L.; Horn, M.A. As the Worm Turns: The Earthworm Gut as a Transient Habitat for Soil Microbial Biomes. Annu. Rev. Microbiol. 2007, 61, 169–189. [Google Scholar] [CrossRef]
- Sampedro, L.; Jeannotte, R.; Whalen, J.K. Trophic transfer of fatty acids from gut microbiota to the earthworm Lumbricus terrestris L. Soil Biol. Biochem. 2006, 38, 2188–2198. [Google Scholar] [CrossRef]
- Wüst, P.K.; Horn, M.A.; Drake, H.L. Clostridiaceae and Enterobacteriaceae as active fermenters in earthworm gut content. ISME J. 2011, 5, 92–106. [Google Scholar] [CrossRef]
- Zeibich, L.; Schmidt, O.; Drake, H.L. Fermenters in the earthworm gut: Do transients matter? FEMS Microbiol. Ecol. 2019, 95, fiy221. [Google Scholar] [CrossRef]
- Furlong, M.A.; Singleton, D.R.; Coleman, D.C.; Whitman, W.B. Molecular and culture-based analyses of prokaryotic communities from an agricultural soil and the burrows and casts of the earthworm Lumbricus rubellus. Appl. Environ. Microbiol. 2002, 68, 1265–1279. [Google Scholar] [CrossRef]
- Singleton, D.R.; Hendrix, P.F.; Coleman, D.C.; Whitman, W.B. Identification of uncultured bacteria tightly associated with the intestine of the earthworm Lumbricus rubellus (Lumbricidae, Oligochaeta). Soil Biol. Biochem. 2003, 35, 1547–1555. [Google Scholar] [CrossRef]
- Knapp, B.A.; Podmirseg, S.M.; Seeber, J.; Meyer, E.; Insam, H. Diet-related composition of the gut microbiota of Lumbricus rubellus as revealed by a molecular fingerprinting technique and cloning. Soil Biol. Biochem. 2009, 41, 2299–2307. [Google Scholar] [CrossRef]
- Egert, M.; Marhan, S.; Wagner, B.; Scheu, S.; Friedrich, M.W. Molecular profiling of 16S rRNA genes reveals diet-related differences of microbial communities in soil, gut, and casts of Lumbricus terrestris L. (Oligochaeta: Lumbricidae). FEMS Microbiol. Ecol. 2004, 48, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Thakuria, D.; Schmidt, O.; Finan, D.; Egan, D.; Doohan, F.M. Gut wall bacteria of earthworms: A natural selection process. ISME J. 2009, 4, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Brandón, M.; Aira, M.; Lores, M.; Domínguez, J. Epigeic Earthworms Exert a Bottleneck Effect on Microbial Communities through Gut Associated Processes. PLoS ONE 2011, 6, e24786. [Google Scholar] [CrossRef]
- Gómez-Brandón, M.; Lores, M.; Domínguez, J. Species-specific effects of epigeic earthworms on microbial community structure during first stages of decomposition of organic matter. PLoS ONE 2012, 7, e31895. [Google Scholar] [CrossRef]
- Aira, M.; Bybee, S.; Pérez-Losada, M.; Domínguez, J. Feeding on microbiomes: Effects of detritivory on the taxonomic and phylogenetic bacterial composition of animal manures. FEMS Microbiol. Ecol. 2015, 91, fiv117. [Google Scholar] [CrossRef]
- Aira, M.; Olcina, J.; Pérez-Losada, M.; Domínguez, J. Characterization of the bacterial communities of casts from Eisenia andrei fed with different substrates. Appl. Soil Ecol. 2016, 98, 103–111. [Google Scholar] [CrossRef]
- Domínguez, J.; Aira, M.; Crandall, K.A.; Pérez-Losada, M. Earthworms drastically change fungal and bacterial communities during vermicomposting of sewage sludge. Sci. Rep. 2021, 11, 15556. [Google Scholar] [CrossRef]
- Aira, M.; Domínguez, J. Earthworm effects without earthworms: Inoculation of raw organic matter with worm-worked substrates alters microbial community functioning. PLoS ONE 2011, 6, e16354. [Google Scholar] [CrossRef]
- Aira, M.; Monroy, F.; Domínguez, J. Ageing effects on nitrogen dynamics and enzyme activities in casts of Aporrectodea caliginosa (Lumbricidae). Pedobiologia 2005, 49, 467–473. [Google Scholar] [CrossRef]
- Aira, M.; Lazcano, C.; Gómez-Brandón, M.; Domínguez, J. Ageing effects of casts of Aporrectodea caliginosa on soil microbial community structure and activity. Appl. Soil Ecol. 2010, 46, 143–146. [Google Scholar] [CrossRef]
- Aira, M.; Pérez-Losada, M.; Domínguez, J. Microbiome dynamics during cast ageing in the earthworm Aporrectodea caliginosa. Appl. Soil Ecol. 2019, 139, 56–63. [Google Scholar] [CrossRef]
- Monroy, F.; Aira, M.; Domínguez, J.; Mariño, F. Distribution of earthworms in the north-west of the Iberian Peninsula. Eur. J. Soil Biol. 2003, 39, 13–18. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the Miseq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in mark-er-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Foster, Z.S.L.; Sharpton, T.J.; Gründwald, N.J. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol. 2017, 13, e1005404. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D. Evaluating different approaches that test whether microbial communities have the same structure. ISME J. 2008, 2, 265–275. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- McCullagh, P.; Nelder, J.A. Generalized Linear Models; Chapman and Hall: London, UK, 1989. [Google Scholar]
- Herrera, C.M. Flower-to-seedling consequences of different pollination regimes in an insect-pollinated shrub. Ecology 2000, 81, 15–29. [Google Scholar] [CrossRef]
- Hothorn, T.; Bretz, F.; Westfall, P. Simultaneous Inference in General Parametric Models. Biom. J. 2008, 50, 346–363. [Google Scholar] [CrossRef]
- Holmes, I.; Harris, K.; Quince, C. Dirichlet multinomial mixtures: Generative models for microbial metagenomics. PLoS ONE 2012, 7, e30126. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, platform-Independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Schloss, P.D. Dynamics and associations of microbial community types across the human body. Nature 2014, 509, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Lian, B.; Wu, C.; GUo, P. A comparative study of gut microbiota profiles of earthworms fed in three different substrates. Symbiosis 2018, 74, 21–29. [Google Scholar] [CrossRef]
- Pass, D.A.; Morgan, A.J.; Read, D.S.; Field, D.; Weightman, A.J.; Kille, P. The effect of anthropogenic arsenic contamination on the earthworm microbiome. Environ. Microb. 2015, 17, 1884–1896. [Google Scholar] [CrossRef] [PubMed]
- Zeibich, L.; Gulh, J.; Drake, H.L. Impact of water content and dietary organic carbon richness on gut bacteria in the earthworm Lumbricus terrestris. FEMS Microbes 2020, 1, xtaa002. [Google Scholar] [CrossRef]
- Wüst, P.K.; Horn, M.A.; Henderson, G.; Janssen, P.H.; Rehm, B.H.A.; Drake, H.L. Gut-associated denitrification and in vivo emission of nitrous oxide by the earthworm families Megascolecidae and Lumbricidae in New Zealand. Appl. Environ. Microbiol. 2009, 75, 3430–3436. [Google Scholar] [CrossRef]
- Sapkova, R.; Santos, S.; Farias, P.; Krogh, P.H.; Winding, A. Insights into the earthworm gut multi-kingdom microbial communities. Sci. Tot. Environ. 2020, 727, 138301. [Google Scholar] [CrossRef]
- Berg, M.; Stenuit, B.; Ho, J.; Wang, A.; Parke, C.; Knight, M.; Alvarez-Cohen, L.; Shapira, M. Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME J. 2016, 10, 1998–2009. [Google Scholar] [CrossRef]
- Chandler, J.A.; Lang, J.M.; Bhatnagar, S.; Eisen, J.A.; Kopp, A. Bacterial Communities of Diverse Drosophila Species: Ecological Context of a Host–Microbe Model System. PLoS Genet. 2011, 7, e1002272. [Google Scholar] [CrossRef]
- Dirksen, P.; Marsh, S.A.; Braker, I.; Heitland, N.; Wagner, S.; Nakad, R.; Mader, S.; Petersen, C.; Kowallik, V.; Rosenstiel, P.; et al. The native microbiome of the nematode Caenorhabditis elegans: Gateway to a new host-microbiome model. BMC Biol. 2016, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Aira, M.; Pérez-Losada, M.; Domínguez, J. Diversity, structure and sources of bacterial communities in earthworm cocoons. Sci. Rep. 2018, 8, 6632. [Google Scholar] [CrossRef] [PubMed]
- Reveillaud, J.; Maignien, L.; Murat Eren, A.; Huber, J.A.; Apprill, A.; Sogin, M.L.; Vanreusel, A. Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J. 2014, 8, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Nechitaylo, T.Y.; Timmis, K.N.; Golyshin, P.N. ‘Candidatus Lumbricincola’, a novel lineage of uncultured Mollicutes from earthworms of family Lumbricidae. Environ. Microbiol. 2009, 11, 1016–1026. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aira, M.; Pérez-Losada, M.; Crandall, K.A.; Domínguez, J. Composition, Structure and Diversity of Soil Bacterial Communities before, during and after Transit through the Gut of the Earthworm Aporrectodea caliginosa. Microorganisms 2022, 10, 1025. https://doi.org/10.3390/microorganisms10051025
Aira M, Pérez-Losada M, Crandall KA, Domínguez J. Composition, Structure and Diversity of Soil Bacterial Communities before, during and after Transit through the Gut of the Earthworm Aporrectodea caliginosa. Microorganisms. 2022; 10(5):1025. https://doi.org/10.3390/microorganisms10051025
Chicago/Turabian StyleAira, Manuel, Marcos Pérez-Losada, Keith A. Crandall, and Jorge Domínguez. 2022. "Composition, Structure and Diversity of Soil Bacterial Communities before, during and after Transit through the Gut of the Earthworm Aporrectodea caliginosa" Microorganisms 10, no. 5: 1025. https://doi.org/10.3390/microorganisms10051025
APA StyleAira, M., Pérez-Losada, M., Crandall, K. A., & Domínguez, J. (2022). Composition, Structure and Diversity of Soil Bacterial Communities before, during and after Transit through the Gut of the Earthworm Aporrectodea caliginosa. Microorganisms, 10(5), 1025. https://doi.org/10.3390/microorganisms10051025