Microbial Community Structure and Ecological Networks during Simulation of Diatom Sinking

Abstract
:1. Introduction
2. Materials and Methods
2.1. Seawater Sampling
2.2. DNA-SIP Experiments
2.3. DNA Extraction and SIP Ultracentrifugation
2.4. Quantitative PCR
2.5. PCR Amplification of 16S rRNA Gene
2.6. Illumina MiSeq Sequencing and Data Processing
2.7. Statistical and Ecological Analyses
2.8. Ecological Network Analysis
3. Results
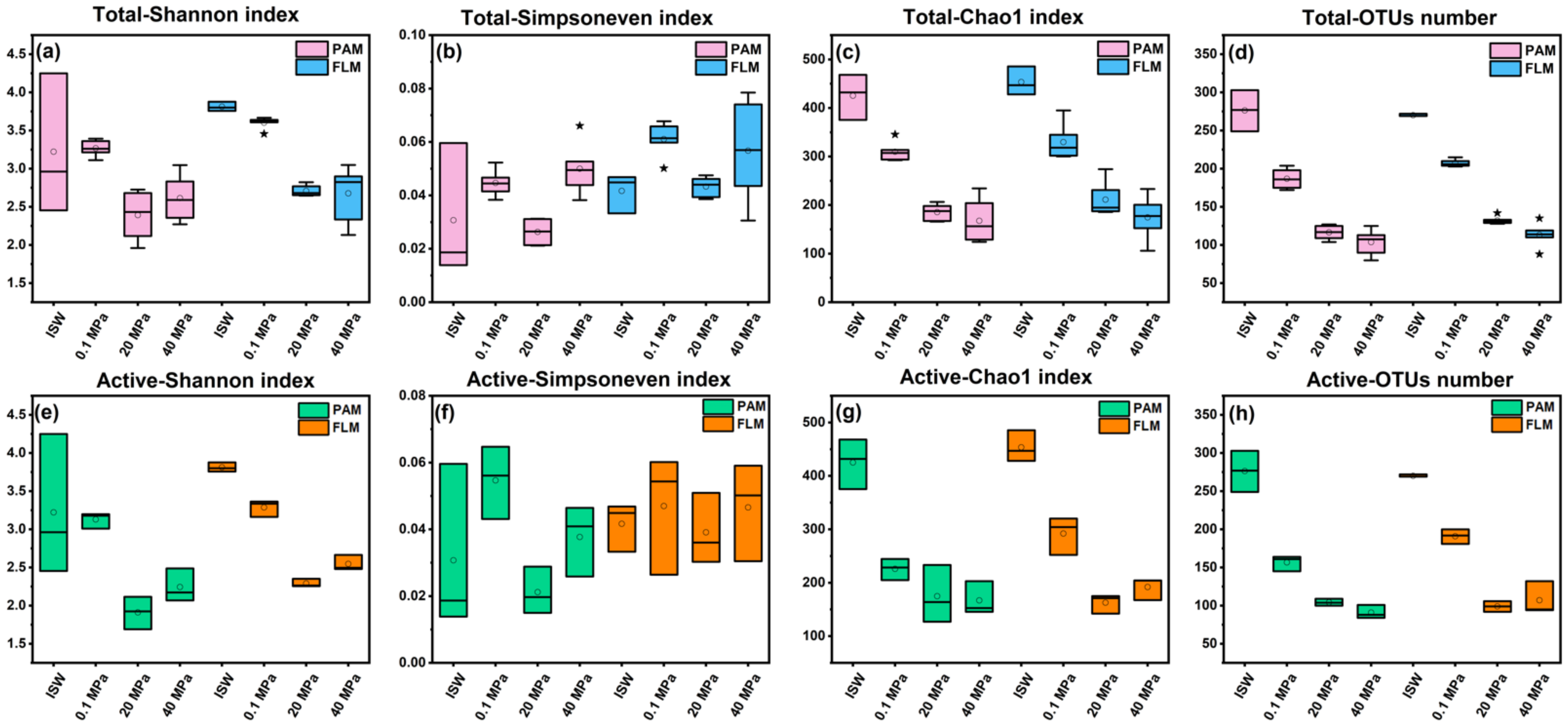
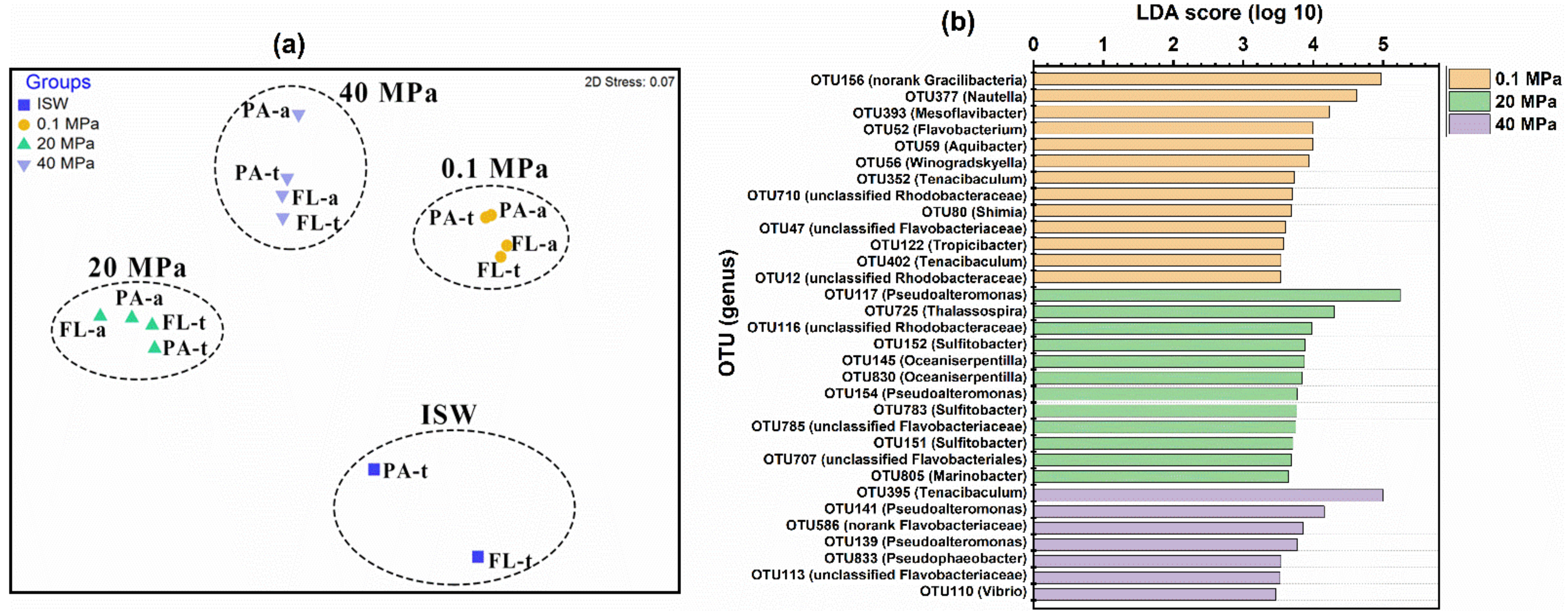
3.1. Microbial Diversity
3.2. Taxonomic Compositions of the PAM and FLM Communities
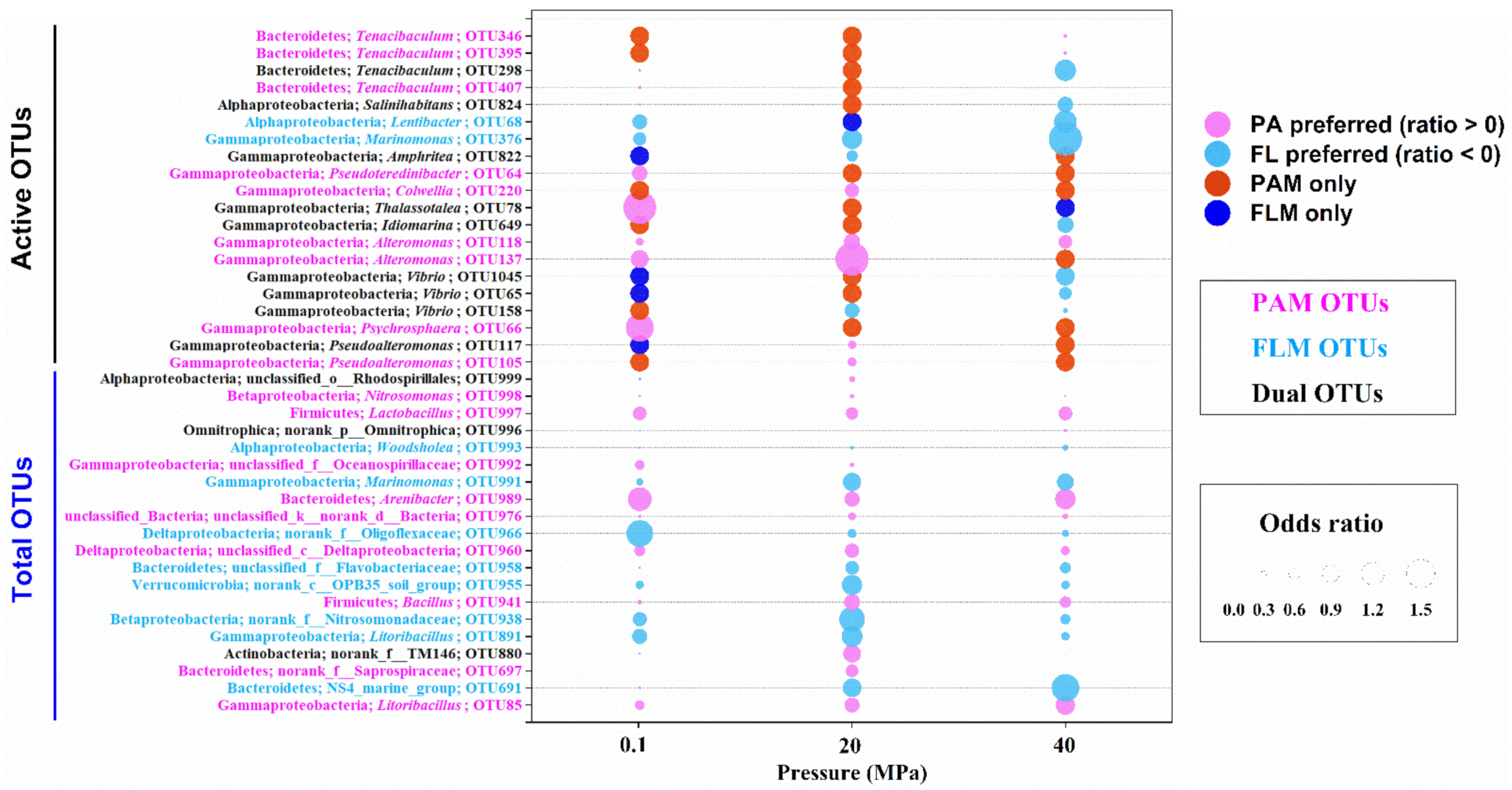
3.3. Microbial Preference for PA or FL Lifestyles
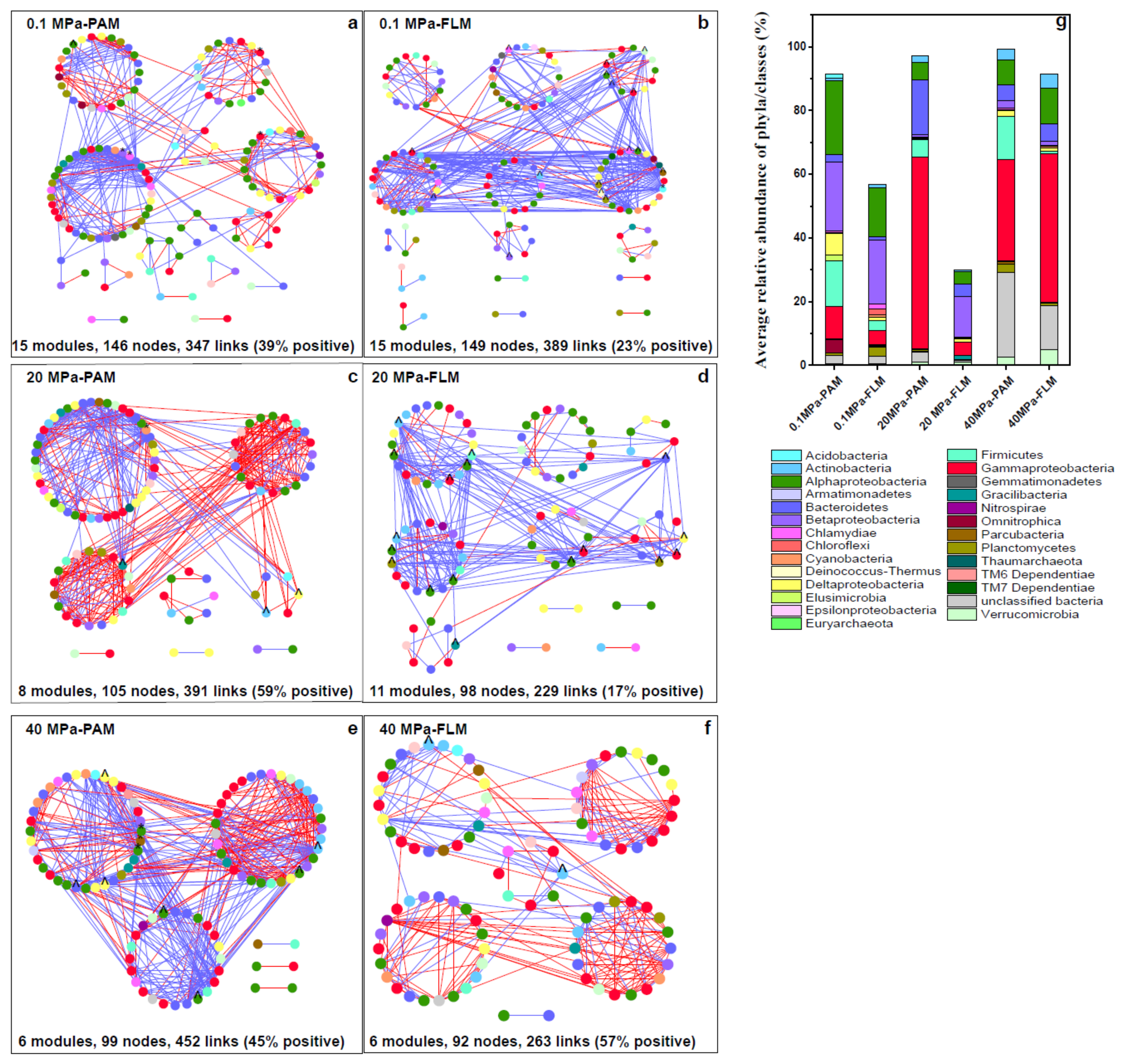
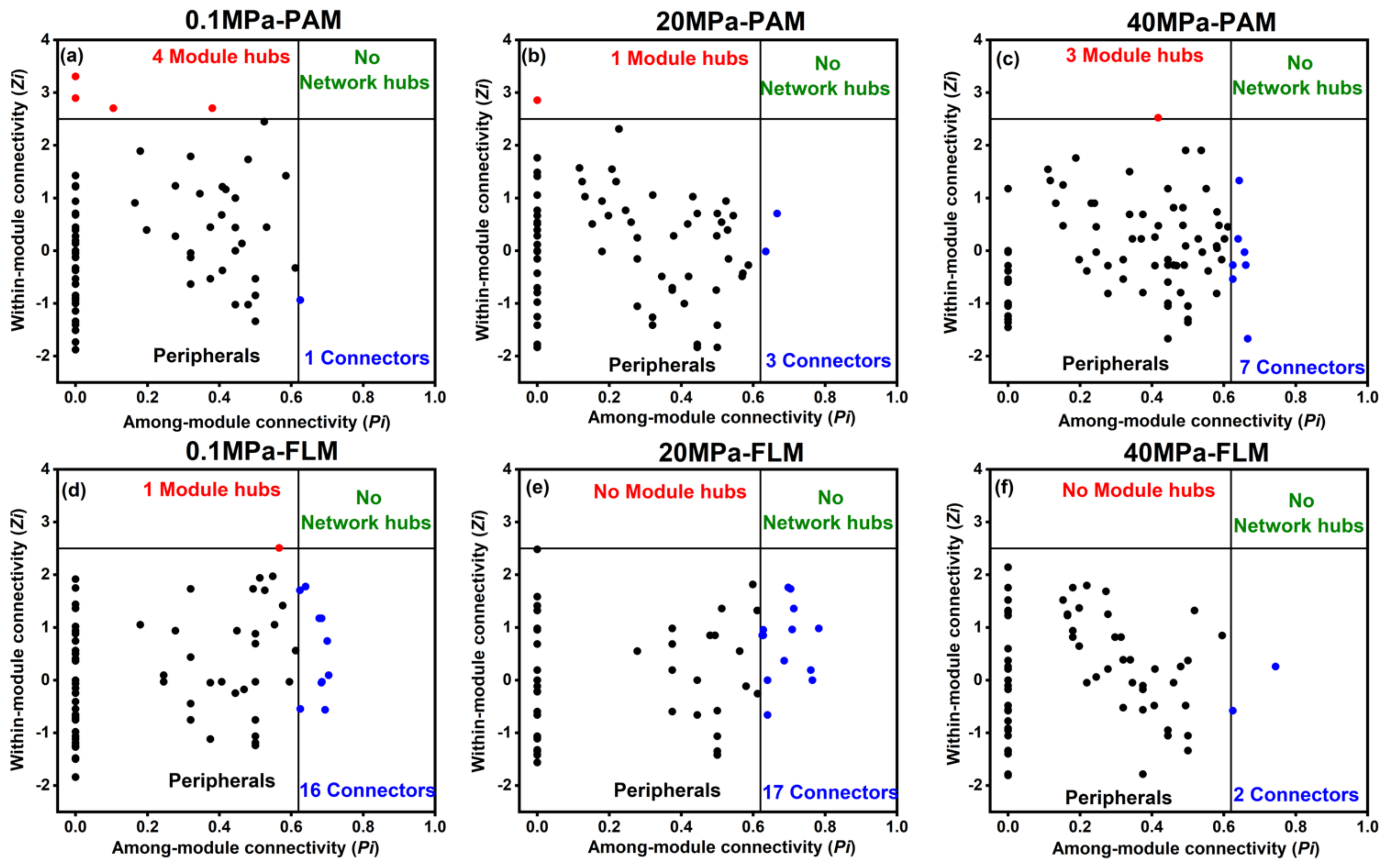
3.4. Ecological Networks
4. Discussion
4.1. Microbial Succession and Lifestyle Preferences during POM Sinking
4.2. Microbial Network Interactions during POM Sinking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 2007, 5, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Arístegui, J.; Gasol, J.M.; Duarte, C.M.; Herndld, G.J. Microbial oceanography of the dark ocean’s pelagic realm. Limnol. Oceanogr. 2009, 54, 1501–1529. [Google Scholar] [CrossRef] [Green Version]
- Herndl, G.J.; Reinthaler, T. Microbial control of the dark end of the biological pump. Nat. Geosci. 2013, 6, 718–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Zhang, L.; Li, J.; Kato, C.; Tamburini, C.; Zhang, Y.; Dang, H.; Wang, G.; Wang, F.J. The POM-DOM piezophilic microorganism continuum (PDPMC)—The role of piezophilic microorganisms in the global ocean carbon cycle. Sci. China Earth Sci. 2015, 58, 106–115. [Google Scholar] [CrossRef]
- Mestre, M.; Ruiz-González, C.; Logares, R.; Duarte, C.M.; Gasol, J.M.; Sala, M.M. Sinking particles promote vertical connectivity in the ocean microbiome. Proc. Natl. Acad. Sci. USA 2018, 115, E6799–E6807. [Google Scholar] [CrossRef] [Green Version]
- Arnosti, C. Microbial Extracellular Enzymes and the Marine Carbon Cycle. Annu. Rev. Mar. Sci. 2011, 3, 401–425. [Google Scholar] [CrossRef]
- Grossart, H.P.; Tang, K.W.; Kiørboe, T.; Ploug, H. Comparison of cell-specific activity between free-living and attached bacteria using isolates and natural assemblages. FEMS Microbiol. Lett. 2007, 266, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Lauro, F.M.; McDougald, D.; Thomas, T.; Williams, T.J.; Egan, S.; Rice, S.; DeMaere, M.Z.; Ting, L.; Ertan, H.; Johnson, J.; et al. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 15527–15533. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Wang, L.; Liu, Q.; Wang, Z.; Li, Z.; Fang, J.; Zhang, L.; Luo, M. Depth-Resolved Distribution of Particle-Attached and Free-Living Bacterial Communities in the Water Column of the New Britain Trench. Front. Microbiol. 2018, 9, 625. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gu, L.; Bai, S.; Wang, J.; Su, L.; Wei, B.; Zhang, L.; Fang, J. Characterization of particle-associated and free-living bacterial and archaeal communities along the water columns of the South China Sea. Biogeosciences 2021, 18, 113–133. [Google Scholar] [CrossRef]
- Ganesh, S.; Parris, D.J.; DeLong, E.F.; Stewart, F.J. Metagenomic analysis of size-fractionated picoplankton in a marine oxygen minimum zone. ISME J. 2014, 8, 187–211. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.; Grossart, H.-P.; Schweitzer, B.; Ploug, H. Microbial ecology of organic aggregates in aquatic ecosystems. Aquat. Microb. Ecol. 2002, 28, 175–211. [Google Scholar] [CrossRef] [Green Version]
- Ghiglione, J.F.; Mevel, G.; Pujo-Pay, M.; Mousseau, L.; Lebaron, P.; Goutx, M. Diel and seasonal variations in abundance, activity, and community structure of particle-attached and free-living bacteria in NW Mediterranean Sea. Microb. Ecol. 2007, 54, 217–231. [Google Scholar] [CrossRef]
- Kellogg, C.T.E.; Carpenter, S.D.; Renfro, A.A.; Sallon, A.; Michel, C.; Cochran, J.K.; Deming, J.W. Evidence for microbial attenuation of particle flux in the Amundsen Gulf and Beaufort Sea: Elevated hydrolytic enzyme activity on sinking aggregates. Polar Biol. 2011, 34, 2007–2023. [Google Scholar] [CrossRef]
- DeLong, E.F.; Franks, D.G.; Alldredge, A.L. Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol. Oceanogr. 1993, 38, 924–934. [Google Scholar] [CrossRef] [Green Version]
- Eloe, E.A.; Shulse, C.N.; Fadrosh, D.W.; Williamson, S.J.; Allen, E.E.; Bartlett, D.H. Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ. Microbiol. Rep. 2011, 3, 449–458. [Google Scholar] [CrossRef]
- Crespo, B.G.; Pommier, T.; Fernández-Gómez, B.; Pedrós-Alió, C. Taxonomic composition of the particle-attached and free-living bacterial assemblages in the Northwest Mediterranean Sea analyzed by pyrosequencing of the 16S rRNA. Microbiologyopen 2013, 2, 541–552. [Google Scholar] [CrossRef]
- Riemann, L.; Winding, A. Community Dynamics of Free-living and Particle-associated Bacterial Assemblages during aFreshwater Phytoplankton Bloom. Microb. Ecol. 2001, 42, 274–285. [Google Scholar] [CrossRef]
- Ortega-Retuerta, E.; Joux, F.; Jeffrey, W.H.; Ghiglione, J.F. Spatial variability of particle-attached and free-living bacterial diversity in surface waters from the Mackenzie River to the Beaufort Sea (Canadian Arctic). Biogeosciences 2013, 10, 2747–2759. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Fang, J.; Jia, Z.; Chen, S.; Zhang, L.; Gao, W. DNA stable-isotope probing reveals potential key players for microbial decomposition and degradation of diatom-derived marine particulate matter. Microbiologyopen 2020, 9, e1013. [Google Scholar] [CrossRef]
- Ghiglione, J.-F.; Conan, P.; Pujo-Pay, M. Diversity of total and active free-living vs. particle-attached bacteria in the euphotic zone of the NW Mediterranean Sea. FEMS Microbiol. Lett. 2009, 299, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ambrosio, L.; Ziervogel, K.; MacGregor, B.; Teske, A.; Arnosti, C. Composition and enzymatic function of particle-associated and free-living bacteria: A coastal/offshore comparison. ISME J. 2014, 8, 2167–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.; Zeigler Allen, L.; Allen, A.; Herfort, L.; Simon, H. Contrasting genomic properties of free-living and particle-attached microbial assemblages within a coastal ecosystem. Front. Microbiol. 2013, 4, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannoni, S.J.; Cameron Thrash, J.; Temperton, B. Implications of streamlining theory for microbial ecology. ISME J. 2014, 8, 1553–1565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiao, W.; Jiao, N. Linking biochemical properties of particles to particle-attached and free-living bacterial community structure along the particle density gradient from freshwater to open ocean. J. Geophys. Res. Biogeosci. 2016, 121, 2261–2274. [Google Scholar] [CrossRef] [Green Version]
- Milici, M.; Deng, Z.-L.; Tomasch, J.; Decelle, J.; Wos-Oxley, M.L.; Wang, H.; Jáuregui, R.; Plumeier, I.; Giebel, H.-A.; Badewien, T.H.; et al. Co-occurrence Analysis of Microbial Taxa in the Atlantic Ocean Reveals High Connectivity in the Free-Living Bacterioplankton. Front. Microbiol. 2016, 7, 649. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wang, Q.; Simon, P.N.; Liu, J.; Liu, L.; Dai, X.; Zhang, X.; Kuang, J.; Igarashi, Y.; Pan, X.; et al. Distinct Network Interactions in Particle-Associated and Free-Living Bacterial Communities during a Microcystis aeruginosa Bloom in a Plateau Lake. Front. Microbiol. 2017, 8, 202. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, D.; Huang, R.; Cao, X.; Zeng, J.; Yu, Z.; Hooker, K.V.; Hambright, K.D.; Wu, Q.L. Contrasting Network Features between Free-Living and Particle-Attached Bacterial Communities in Taihu Lake. Microb. Ecol. 2018, 76, 303–313. [Google Scholar] [CrossRef]
- Tamburini, C.; Garcin, J.; Grégori, G.; Leblanc, K.; Rimmelin, P.; Kirchman, D.L. Pressure effects on surface Mediterranean prokaryotes and biogenic silica dissolution during a diatom sinking experiment. Aquat. Microb. Ecol. 2006, 43, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Tamburini, C.; Garel, M.; Barani, A.; Boeuf, D.; Bonin, P.; Bhairy, N.; Guasco, S.; Jacquet, S.; Le Moigne, F.A.C.; Panagiotopoulos, C.; et al. Increasing Hydrostatic Pressure Impacts the Prokaryotic Diversity during Emiliania huxleyi Aggregates Degradation. Water 2021, 13, 2616. [Google Scholar] [CrossRef]
- Fang, J.; Zhang, L.; Bazylinski, D.A. Deep-sea piezosphere and piezophiles: Geomicrobiology and biogeochemistry. Trends Microbiol. 2010, 18, 413–422. [Google Scholar] [CrossRef]
- Oger, P.M.; Jebbar, M. The many ways of coping with pressure. Res. Microbiol. 2010, 161, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, X.; Bartlett, D.H.; Xiao, X. Current developments in marine microbiology: High-pressure biotechnology and the genetic engineering of piezophiles. Curr. Opin. Biotechnol. 2015, 33, 157–164. [Google Scholar] [CrossRef]
- Xiao, X.; Zhang, Y.; Wang, F. Hydrostatic pressure is the universal key driver of microbial evolution in the deep ocean and beyond. Environ. Microbiol. Rep. 2021, 13, 68–72. [Google Scholar] [CrossRef]
- Grossart, H.P.; Gust, G.J. Hydrostatic pressure affects physiology and community structure of marine bacteria during settling to 4000 m: An experimental approach. Mar. Ecol. Prog. Ser. 2009, 390, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Marietou, A.; Bartlett, D.H.; Kostka, J.E. Effects of High Hydrostatic Pressure on Coastal Bacterial Community Abundance and Diversity. Appl. Environ. Microbiol. 2014, 80, 5992–6003. [Google Scholar] [CrossRef] [Green Version]
- Tamburini, C.; Boutrif, M.; Garel, M.; Colwell, R.R.; Deming, J.W. Prokaryotic responses to hydrostatic pressure in the ocean—A review. Environ. Microbiol. 2013, 15, 1262–1274. [Google Scholar] [CrossRef]
- Stief, P.; Elvert, M.; Glud, R.N. Respiration by “marine snow” at high hydrostatic pressure: Insights from continuous oxygen measurements in a rotating pressure tank. Limnol. Oceanogr. 2021, 66, 2797–2809. [Google Scholar] [CrossRef]
- Berges, J.A.; Franklin, D.J.; Harrison, P.J. Evolution of an artificial seawater medium: Improvements in enriched seawater, artificial water over the last two decades. J. Phycol. 2001, 37, 1138–1145. [Google Scholar] [CrossRef]
- Pasotti, F.; De Troch, M.; Raes, M.; Vanreusel, A. Feeding ecology of shallow water meiofauna: Insights from a stable isotope tracer experiment in Potter Cove, King George Island, Antarctica. Polar Biol. 2012, 35, 1629–1640. [Google Scholar] [CrossRef]
- Fang, J.; Uhle, M.; Billmark, K.; Bartlett, D.H.; Kato, C. Fractionation of carbon isotopes in biosynthesis of fatty acids by a piezophilic bacterium Moritella japonica strain DSK1. Geochim. Cosmochim. Acta 2006, 70, 1753–1760. [Google Scholar] [CrossRef]
- Neufeld, J.D.; Vohra, J.; Dumont, M.G.; Lueders, T.; Manefield, M.; Friedrich, M.W.; Murrell, J.C. DNA stable-isotope probing. Nat. Protoc. 2007, 2, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Zhang, C.; Zeng, X.; Feng, Y.; Weng, J.; Lin, X.; Zhu, J.; Xiong, Z.; Xu, J.; Cai, Z.; et al. Autotrophic growth of nitrifying community in an agricultural soil. ISME J. 2011, 5, 1226–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijs, S.; Op De Beeck, M.; Beckers, B.; Truyens, S.; Stevens, V.; Van Hamme, J.D.; Weyens, N.; Vangronsveld, J. Comparative Evaluation of Four Bacteria-Specific Primer Pairs for 16S rRNA Gene Surveys. Front. Microbiol. 2017, 8, 494. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. Msystems 2016, 1, e00009–e00015. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2013, 42, D633–D642. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.R.; Gorley, R.N. Getting Started with PRIMER v7. In PRIMER-E: Plymouth; Plymouth Marine Laboratory: Devon, UK, 2015. [Google Scholar]
- Venny Oliveros, J.C. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 27 February 2020).
- Suter, E.A.; Pachiadaki, M.; Taylor, G.T.; Astor, Y.; Edgcomb, V.P. Free-living chemoautotrophic and particle-attached heterotrophic prokaryotes dominate microbial assemblages along a pelagic redox gradient. Environ. Microbiol. 2018, 20, 693–712. [Google Scholar] [CrossRef]
- Orsi, W.D.; Smith, J.M.; Liu, S.; Liu, Z.; Sakamoto, C.M.; Wilken, S.; Poirier, C.; Richards, T.A.; Keeling, P.J.; Worden, A.Z.; et al. Diverse, uncultivated bacteria and archaea underlying the cycling of dissolved protein in the ocean. ISME J. 2016, 10, 2158–2173. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Tu, Q.; Zhi, X.; Relman, D.A. Functional Molecular Ecological Networks. MBio 2010, 1, e00169-10. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Clauset, A.; Newman, M.E.J.; Moore, C. Finding community structure in very large networks. Phys. Rev. E 2004, 70, 66111. [Google Scholar] [CrossRef] [Green Version]
- Guimerà, R.; Nunes Amaral, L.A. Functional cartography of complex metabolic networks. Nature 2005, 433, 895–900. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T.J. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Tu, Q.; Yan, Q.; Deng, Y.; Michaletz, S.T.; Buzzard, V.; Weiser, M.D.; Waide, R.; Ning, D.; Wu, L.; He, Z.; et al. Biogeographic patterns of microbial co-occurrence ecological networks in six American forests. Soil Biol. Biochem. 2020, 148, 107897. [Google Scholar] [CrossRef]
- Wan, X.; Gao, Q.; Zhao, J.; Feng, J.; van Nostrand, J.D.; Yang, Y.; Zhou, J. Biogeographic patterns of microbial association networks in paddy soil within Eastern China. Soil Biol. Biochem. 2020, 142, 107696. [Google Scholar] [CrossRef]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; He, Z.; Zhou, J.; Firestone, M.K. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Li, Y.; Zhang, W.; Niu, L.; Gao, Y.; Hui, C.; Bertilsson, S. Hydrostatic pressure influence activity and assembly of bacterial communities in reservoir sediments. Freshw. Biol. 2021, 66, 1049–1059. [Google Scholar] [CrossRef]
- Jebbar, M.; Franzetti, B.; Girard, E.; Oger, P. Microbial diversity and adaptation to high hydrostatic pressure in deep-sea hydrothermal vents prokaryotes. Extremophiles 2015, 19, 721–740. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Lovell, C.R. Microbial Surface Colonization and Biofilm Development in Marine Environments. Microbiol. Mol. Biol. Rev. 2016, 80, 91–138. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wei, B.; Wang, J.; Liu, Y.; Dasgupta, S.; Zhang, L.; Fang, J. Variation in abundance and community structure of particle-attached and free-living bacteria in the South China Sea. Deep Sea Res. Part II Top. Stud. Oceanogr. 2015, 122, 64–73. [Google Scholar] [CrossRef]
- Fang, J.; Kato, C.; Runko, G.M.; Nogi, Y.; Hori, T.; Li, J.; Morono, Y.; Inagaki, F. Predominance of viable spore-forming piezophilic bacteria in high-pressure enrichment cultures from ~1.5 to 2.4 km-deep coal-bearing sediments below the ocean floor. Front. Microbiol. 2017, 8, 137. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.-L.; Wang, Z.-B.; Su, H.-N.; Chen, X.-L.; Miao, J.; Wang, X.-J.; Li, C.-Y.; Zhang, X.-Y.; Li, P.-Y.; Wang, M.J. Oxidation of trimethylamine to trimethylamine N-oxide facilitates high hydrostatic pressure tolerance in a generalist bacterial lineage. Sci. Adv. 2021, 7, eabf9941. [Google Scholar] [CrossRef]
- Teeling, H.; Fuchs, B.M.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.; Mann, A.J.; Waldmann, J.; et al. Substrate-Controlled Succession of Marine Bacterioplankton Populations Induced by a Phytoplankton Bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef]
- Buchan, A.; LeCleir, G.R.; Gulvik, C.A.; González, J.M. Master recyclers: Features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Microbiol. 2014, 12, 686–698. [Google Scholar] [CrossRef]
- Mayali, X.; Stewart, B.; Mabery, S.; Weber, P.K. Temporal succession in carbon incorporation from macromolecules by particle-attached bacteria in marine microcosms. Environ. Microbiol. Rep. 2016, 8, 68–75. [Google Scholar] [CrossRef]
- Feng, B.-W.; Li, X.-R.; Wang, J.-H.; Hu, Z.-Y.; Meng, H.; Xiang, L.-Y.; Quan, Z.-X. Bacterial diversity of water and sediment in the Changjiang estuary and coastal area of the East China Sea. FEMS Microbiol. Ecol. 2009, 70, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Wu, Y.; Zhu, Z.; Wang, X.; Li, Z.; Zhang, J. Bacterial diversity in the surface sediments of the hypoxic zone near the Changjiang Estuary and in the East China Sea. Microbiologyopen 2016, 5, 323–339. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Zhang, Z.; Sun, M.; Feng, Z.; Ye, M.J. Characterization and ecological function of bacterial communities in seabed sediments of the southwestern Yellow Sea and northwestern East China Sea, Western Pacific. Sci. Total Environ. 2021, 761, 143233. [Google Scholar] [CrossRef]
- McCarren, J.; Becker, J.W.; Repeta, D.J.; Shi, Y.; Young, C.R.; Malmstrom, R.R.; Chisholm, S.W.; DeLong, E.F. Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc. Natl. Acad. Sci. USA 2010, 107, 16420–16427. [Google Scholar] [CrossRef] [Green Version]
- Tada, Y.; Taniguchi, A.; Sato-Takabe, Y.; Hamasaki, K. Growth and succession patterns of major phylogenetic groups of marine bacteria during a mesocosm diatom bloom. J. Oceanogr. 2012, 68, 509–519. [Google Scholar] [CrossRef]
- Taylor, J.D.; Cunliffe, M. Coastal bacterioplankton community response to diatom-derived polysaccharide microgels. Environ. Microbiol. Rep. 2017, 9, 151–157. [Google Scholar] [CrossRef]
- Mönnich, J.; Tebben, J.; Bergemann, J.; Case, R.; Wohlrab, S.; Harder, T. Niche-based assembly of bacterial consortia on the diatom Thalassiosira rotula is stable and reproducible. ISME J. 2020, 14, 1614–1625. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Fang, J.; Li, J.; Zhang, L.; Xie, B.-B.; Chen, X.-L.; Zhang, Y.-Z. Depth-Resolved Variations of Cultivable Bacteria and Their Extracellular Enzymes in the Water Column of the New Britain Trench. Front. Microbiol. 2018, 9, 135. [Google Scholar] [CrossRef]
- Bidle, K.D.; Azam, F. Bacterial control of silicon regeneration from diatom detritus: Significance of bacterial ectohydrolases and species identity. Limnol. Oceanogr. 2001, 46, 1606–1623. [Google Scholar] [CrossRef]
- Rehnstam-Holm, A.-S.; Godhe, A.; Härnström, K.; Raghunath, P.; Saravanan, V.; Collin, B.; Karunasagar, I.; Karunasagar, I. Association between phytoplankton and Vibrio spp. along the southwest coast of India: A mesocosm experiment. Aquat. Microb. Ecol. 2010, 58, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Frischkorn, K.R.; Stojanovski, A.; Paranjpye, R. Vibrio parahaemolyticus type IV pili mediate interactions with diatom-derived chitin and point to an unexplored mechanism of environmental persistence. Environ. Microbiol. 2013, 15, 1416–1427. [Google Scholar] [CrossRef]
- Vojvoda, J.; Lamy, D.; Sintes, E.; Garcia, J.A.; Turk, V.; Herndl, G.J.J. Seasonal variation in marine-snow-associated and ambient-water prokaryotic communities in the northern Adriatic Sea. Aquat. Microb. Ecol. 2014, 73, 211–224. [Google Scholar] [CrossRef] [Green Version]
- Kirchman, D.L. The ecology of Cytophaga—Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 2002, 39, 91–100. [Google Scholar] [CrossRef]
- Bauer, M.; Kube, M.; Teeling, H.; Richter, M.; Lombardot, T.; Allers, E.; Würdemann, C.A.; Quast, C.; Kuhl, H.; Knaust, F.J. Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 2006, 8, 2201–2213. [Google Scholar] [CrossRef]
- Fernández-Gómez, B.; Richter, M.; Schüler, M.; Pinhassi, J.; Acinas, S.G.; González, J.M.; Pedrós-Alió, C. Ecology of marine Bacteroidetes: A comparative genomics approach. ISME J. 2013, 7, 1026–1037. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.J.; Wilkins, D.; Long, E.; Evans, F.; DeMaere, M.Z.; Raftery, M.J.; Cavicchioli, R. The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ. Microbiol. 2013, 15, 1302–1317. [Google Scholar] [CrossRef]
- Grossart, H.P. Ecological consequences of bacterioplankton lifestyles: Changes in concepts are needed. Environ. Microbiol. Rep. 2010, 2, 706–714. [Google Scholar] [CrossRef]
- Simon, H.M.; Smith, M.W.; Herfort, L. Metagenomic insights into particles and their associated microbiota in a coastal margin ecosystem. Front. Microbiol. 2014, 5, 466. [Google Scholar] [CrossRef]
- Salazar, G.; Cornejo-Castillo, F.M.; Borrull, E.; Díez-Vives, C.; Lara, E.; Vaqué, D.; Arrieta, J.M.; Duarte, C.M.; Gasol, J.M.; Acinas, S.G. Particle-association lifestyle is a phylogenetically conserved trait in bathypelagic prokaryotes. Mol. Ecol. 2015, 24, 5692–5706. [Google Scholar] [CrossRef]
- Azam, F.; Long, R.A. Sea snow microcosms. Nature 2001, 414, 495–498. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.; Yu, J.; Feng, K.; Deng, Y.; Zhang, Y.J. The Coexistence Relationship Between Plants and Soil Bacteria Based on Interdomain Ecological Network Analysis. Front. Microbiol. 2021, 12, 745582. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Hayakawa, D.H.; Tripp, H.J.; Stingl, U.; Givan, S.A.; Cho, J.C.; Oh, H.M.; Kitner, J.B.; Vergin, K.L.; Rappé, M.S. The small genome of an abundant coastal ocean methylotroph. Environ. Microbiol. 2008, 10, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, B.; Dann, L.M.; Mitchell, J.G.; Hu, X.; Tang, H.; Zhang, H.; Sheng, Y.J. Bacterial community structure in the Bohai Strait provides insights into organic matter niche partitioning. Cont. Shelf Res. 2018, 169, 46–54. [Google Scholar] [CrossRef]
- Sowell, S.M.; Abraham, P.E.; Shah, M.; Verberkmoes, N.C.; Smith, D.P.; Barofsky, D.F.; Giovannoni, S.J. Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J. 2011, 5, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Ezzeline, J.A.; Desdevises, Y.; Jacquet, S.J. Exploring archaeal and bacterial diversity and co-occurrence in Lake Geneva. Adv. Oceanogr. Limnol. 2020, 11, 2. Available online: https://pagepressjournals.org/index.php/aiol/article/view/aiol.2020.9099 (accessed on 15 December 2021).
- Sieber, C.M.; Paul, B.G.; Castelle, C.J.; Hu, P.; Tringe, S.G.; Valentine, D.L.; Andersen, G.L.; Banfield, J.F.J. Unusual metabolism and hypervariation in the genome of a gracilibacterium (Bd1-5) from an oil-degrading community. MBio 2019, 10, e02128-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. MBio 2011, 2, e00122-11. [Google Scholar] [CrossRef] [Green Version]
- Lupatini, M.; Suleiman, A.K.A.; Jacques, R.J.S.; Antoniolli, Z.I.; de Siqueira Ferreira, A.; Kuramae, E.E.; Roesch, L.F. Network topology reveals high connectance levels and few key microbial genera within soils. Front. Environ. Sci. 2014, 2, 10. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0 | Network Features | 0.1 MPa-PAM (Threshold Value = 0.94) | 20 MPa-PAM (0.91) | 40 MPa-PAM (0.91) | 0.1 MPa-FLM (0.95) | 20 MPa-FLM (0.93) | 40 MPa-FLM (0.93) |
|---|---|---|---|---|---|---|---|
| Empirical networks | Total OTUs | 289 | 166 | 181 | 297 | 196 | 187 |
| Total nodes | 146 | 105 | 99 | 149 | 98 | 92 | |
| Total links | 347 | 391 | 452 | 389 | 229 | 263 | |
| Total positive links | 136 | 230 | 204 | 88 | 40 | 151 | |
| Total negative links | 211 | 161 | 248 | 301 | 189 | 112 | |
| Total modules | 15 | 8 | 6 | 15 | 11 | 6 | |
| Modularity | 0.60 | 0.52 | 0.35 | 0.49 | 0.40 | 0.60 | |
| Average connectivity (avgK) | 4.75 | 7.45 | 9.13 | 5.22 | 4.67 | 5.72 | |
| Average clustering coefficient (avgCC) | 0.27 | 0.32 | 0.37 | 0.17 | 0.12 | 0.28 | |
| Average geodesic distance (GD) | 4.24 | 3.46 | 2.57 | 4.23 | 5.56 | 3.52 | |
| Random networks | Average clustering coefficient (avgCC) | 0.06 ± 0.01 | 0.12 ± 0.01 | 0.16 ± 0.02 | 0.08 ± 0.01 | 0.11 ± 0.02 | 0.08 ± 0.01 |
| Average geodesic distance (GD) | 3.24 ± 0.05 | 2.64 ± 0.04 | 2.40 ± 0.03 | 3.11 ± 0.05 | 3.04 ± 0.08 | 2.82 ± 0.04 | |
| Modularity | 0.41 ± 0.01 | 0.28 ± 0.01 | 0.24 ± 0.01 | 0.37 ± 0.01 | 0.37 ± 0.01 | 0.34 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Zeng, M.; Xie, Z.; Ning, D.; Zhou, J.; Yu, X.; Liu, R.; Zhang, L.; Fang, J. Microbial Community Structure and Ecological Networks during Simulation of Diatom Sinking. Microorganisms 2022, 10, 639. https://doi.org/10.3390/microorganisms10030639
Liu Y, Zeng M, Xie Z, Ning D, Zhou J, Yu X, Liu R, Zhang L, Fang J. Microbial Community Structure and Ecological Networks during Simulation of Diatom Sinking. Microorganisms. 2022; 10(3):639. https://doi.org/10.3390/microorganisms10030639
Chicago/Turabian StyleLiu, Ying, Mengchu Zeng, Zhe Xie, Daliang Ning, Jizhong Zhou, Xi Yu, Rulong Liu, Li Zhang, and Jiasong Fang. 2022. "Microbial Community Structure and Ecological Networks during Simulation of Diatom Sinking" Microorganisms 10, no. 3: 639. https://doi.org/10.3390/microorganisms10030639
APA StyleLiu, Y., Zeng, M., Xie, Z., Ning, D., Zhou, J., Yu, X., Liu, R., Zhang, L., & Fang, J. (2022). Microbial Community Structure and Ecological Networks during Simulation of Diatom Sinking. Microorganisms, 10(3), 639. https://doi.org/10.3390/microorganisms10030639